Abstract
India’s population complexity presents varied challenges in genetic research, and while facilities have gained traction in tier-1 and -2 cities, reliance on international collaborations often delays such investigations. COVID-19 further exacerbated the issues with such sample sharing. Congenital Hyperinsulinism (CHI) is a rare genetic disorder of pancreatic β-cells causing hypoglycaemia in children due to abnormal insulin secretion. Given India’s high birth rate and consanguineous populations, annual CHI cases are estimated to be around up to 10,000, with up to 50% having unexplained genetic causes. Diffuse or atypical lesions in such patients often necessitate near-total-pancreatectomy, risking pancreatic exocrine insufficiency and diabetes, requiring lifelong therapy. Also, novel genetic variations complicate accurate diagnosis, risk assessment, and counselling, emphasising the need for rapid genetic assessment to prevent neurological injuries and inform treatment decisions. Despite significant efforts at many institutes, there are no dedicated organisations for CHI in India. With the implementation of the National Policy for Rare Diseases 2021, we plan to form a non-profit organisation, “Congenital Hyperinsulinism India Association (CHIA)”, comprising paediatric endocrinologists, paediatricians, geneticists, and independent researchers. The aims of this association are to generate a national database registry of patients, formulate a parent support group and CHIA consortium, design patient information leaflets, as well as foster genomic collaborations and promote clinical trials. Such steps will help sensitise the health authorities and policy makers, urging them to improve the allocation of health budgets for rare diseases, as well as empower patients and their families, contributing towards a better quality of life.
1. Introduction
1.1. Scenario of Genetic Diseases in India
India holds the distinction of being the most populous country in the world with 1.44 billion people, representing 17.5% of the global population. The population is also expected to increase at an annual rate of 1.0 percent from 2011 (1.21 billion) to 2036 (1.51 billion), currently standing at 1.39 billion [1]. The country is believed to have been inhabited by migrants from the African subcontinent approximately 65,000 years ago, followed by numerous events of migration and invasion causing a population admixture that has led to highly heterogeneous population groups [2,3,4,5]. At present, there are over 4000 anthropologically distinct groups and 21 different languages with various dialects in India. Recently, the Indian Genome variation consortium identified polymorphism in 900 genes from 55 different population groups of India that are now catalogued in the Indian Genome Variation Browser [6]. With such vast geographical and multi-linguistic population in a socially and culturally deep-rooted religious/caste system, the genetic make-up of the population is further complicated by the high degree of endogamy, with consanguineous marriages as common as 20–30% in specific population groups [7,8]. This places India in a unique position, with the possibility of clusters of specific diseases and founder mutations [9].
The National Health Policy, 2017 of India envisaged an increase in expenditure of 2.5% of the GDP (Gross Domestic Product) by 2025, while the central and state government’s budgeted expenditure on the health sector reached 2.1% in 2021–22 against 1.3% in 2019–20 [10]. Currently, India spends 3.73% of its GDP on its health budget [11], focusing on services providing free and subsidised public health services as executed by all 49 state governments, provided by the Government of India under the Ministry of Health and Family Welfare (https://pib.gov.in/PressNoteDetails.aspx?NoteId=153237&ModuleId=3®=3&lang=1 accessed on 24 October 2024). These are accessed by 25–30% of the overall population, with the rest of the population being served by heterogeneous private health services managed by various corporate organisations and individual practices. Here, geographic and economic disparity plays a pivotal role in the availability of infrastructural resources between the urban and rural population, as well as the economically privileged and deprived classes [12,13].
Such factors pose a considerable challenge for genetic studies and genomic medicine research in India. However, the growing significance of molecular genetics and cytogenetics in diagnosis, as well as the management of many disorders, have led to state-of-the-art molecular diagnostic and counselling facilities in India. Most of the genetic services in terms of clinical as well as molecular geneticists, genomic laboratories and counselling infrastructure are available at tier-1 and -2 cities and premier institutes, while their availability across tier-3 and -4 cities, as well as rural areas, is much lower. Almost all genetic disorders have been reported in India. While such research often comes from a few institutions and individuals, there is a need to establish nationwide prevalence and create a registry of rare genetic disorders. This would also enable the genetic evaluation of a majority of the population and develop a national public health programme for carrier or newborn screening for genetic disorders, as well as advance research in understanding the pathogenesis and discovering newer treatment modalities for genetic disorders and in areas like gene mapping and gene and stem cell therapy [14,15,16].
In the past, genetic investigations have primarily been conducted outside the country on a peer-to-peer basis, with individual investigators coordinating sample collection, storage, maintenance, and redistribution. This has resulted in a challenging turn-around time, often consuming the critical period of a serious illness. And as the community demands have risen, such commitments have become increasingly burdensome, costly, and legally and ethically complex. Recently, COVID-19 laid bare the challenges of sharing clinical and other biological samples. Following the pandemic, there is a demand for the wider adoption of electronic legal agreements for sample transfer and increased harmonisation, interoperability, and searching capacity for sample collection. These issues are unlikely to go away, unless there is more support and stronger incentives from funders and publishers for infrastructure; meticulous and organised utilisation of the available resources and current efforts, which work in silos; and their unification into a national cause, particularly in the context of rare genetic disorders [17].
1.2. Congenital Hyperinsulinism
Congenital Hyperinsulinism (CHI) is a rare genetic disorder of the pancreatic β-cells known to cause recurring episodes of hypoglycaemia due to dysregulated insulin secretion, leading to dangerously low blood glucose concentrations in infants and children [18]. Such inappropriate insulin secretion can also suppress the production of ketone bodies, which serve as an alternative fuel for the brain during hypoglycaemia. This lack of glucose and the deprivation of alternative fuels increase the risk of brain damage in these patients, causing an increase in long-term morbidity as well as mortality [19,20]. With an annual birth rate of 25 million children, India accounts for nearly one-fifth of the world’s annual child births. Up to 1.7 million newborn babies are estimated to be born with birth defects/genetic conditions, with a report supporting 9% of newborns have birth defects [21]. This accounts for at least 625 children estimated to be born with CHI annually, at an incidence of 1 in 40,000 live births in the general population, and this could go up to 10,000 per year, with an incidence of 1 in 2500 in populations with high rates of consanguinity [22,23]. The associated genetic loci causing monogenic forms have been identified as ABCC8 and KCNJ11 [24,25,26,27,28,29], GLUD1 [30,31], GCK [32], HADH1 [33], SLC16A1 [34,35], INSR [36,37], UCP2 [38,39], HNF4α and HNF1α [40,41,42,43,44,45], and HK1 [46]. More than over 20 syndromic forms of CHI have reported so far with multi-systemic involvement, the commonly reported ones being KCNQ1/CDKN1C (Beckwith–Wiedemann syndrome) and KMT2D/KDM6A (Kabuki Syndrome), amongst other syndromic forms [47,48,49,50,51,52,53]. However, despite significant advances which have expanded our understanding of the pathophysiology of this disorder, the cause of CHI remains unknown in up to 50% of, patients suggesting the existence of additional genetic loci that have yet to be discovered [54]. Moreover, up to one-third to one-half of KATP channel mutations detected by genetic testing are novel variants that are interpreted as variants of unknown significance [55]. The optimal management of hyperinsulinism is dependent upon the underlying cause, and it highlights the importance of a good turn-around time of the genetic evaluation, which is also essential to prevent neuroglycopenic brain damage. Also, the characterisation of novel variants and their associated phenotypes is essential for the accurate diagnosis of affected children, improving the interpretation of genetic tests, counselling families about recurrence risks, and identifying other family members at risk of hypoglycaemia [56].
Histologically, CHI has been classified as either a focal form with the involvement of a small area of islet cell expansion and minimal exocrine tissue involvement or a diffuse form involving the entire pancreas and nucleomegaly of some islet cells or atypical forms with a combination of both focal and diffuse lesions [57] (Figure 1). While the management of CHI includes nutritional, medical, and surgical intervention depending on the underlying histologic and genetic subtype, focal lesionectomy is the preferred curative treatment for focal CHI when a complete resection is achieved. But the management of diffuse lesions, which account for the majority of the cases, still poses a significant challenge with near-total-pancreatectomy, i.e., resection of 95–98% of pancreatic tissue as a last resort in preventing hypoglycaemic brain damage, especially in severe cases where sufficient glycaemic control cannot be achieved, even though the combination of different medications and nutritional supplements poses a persistent risk of hypoglycaemia and subsequent neurodevelopmental delay [58,59,60]. However, even surgical interventions with near-total-pancreatectomy have been proven to be not curative in such patients, with often inconsistent and unsatisfactory outcomes, ranging from persisting hypoglycaemia (up to 60%), hyperglycaemia (almost 100% at 11 years post-surgery), and exocrine pancreatic insufficiency (almost 50%) [61]. Moreover, almost 96% of the patients reported the incidence of insulin-dependent diabetes mellitus 10–15 years after near-total-pancreatectomy, 77% after 7 years, and 13% soon after surgery [57].
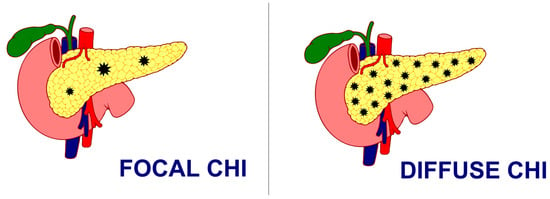
Figure 1.
Focal CHI and Diffuse CHI.
It is very important to follow-up children with this condition in view of some genetic forms that can lead to diabetes later in life. Also, children with pancreatectomy are at a higher risk of developing pancreatic exocrine insufficiency and diabetes later in life.
1.3. Indian Organisations for CHI
With significant advances in gene mapping achieved through molecular genetics and exome sequencing, there exists substantial research on CHI in India, especially from leading institutes like the All India Institute of Medical Sciences, New Delhi; Postgraduate Institute of Medical Education and Research, Chandigarh; Madras Diabetic Research Foundation, Chennai; amongst many others. There has also been a significant amount of work on CHI across the country that includes prominent clinicians in government and private institutions. However, while there are various organisations in India working in the field of genetic disorders, as well as rare diseases [62], our literature review did not find any particular organisation working for CHI in India. With the implementation of National Policy for Rare Disease (NPRD), 2021 by the Ministry of Health and Family Welfare, the Government of India aims to lower the incidence and prevalence of rare diseases based on an integrated and comprehensive preventive strategy [63]. Thus, we plan to form the Congenital Hyperinsulinism India Association (CHIA).
1.4. Global Organisations for CHI
- ○
- Congenital Hyperinsulinism International (https://congenitalhi.org)–Congenital Hyperinsulinism International is a grass-root level organisation founded in 2005 by the concerned parents of children with hyperinsulinism. It is a global organisation supported in its work by a scientific advisory team comprising eleven leading hyperinsulinism world specialists. It remains dedicated to increasing awareness; providing education, information, and support to those living with the disease; advocating on behalf of the patients for better treatments and access to care; and improving the lives of the babies, children, and adults affected by the disorder. It also supports medical research for improved therapies, potential cures, and timely diagnosis.
- ○
- Congenital Hyperinsulinism Charity UK (CHC-UK).
- ○
- There are also family support groups reported in Germany, Spain, and various other countries.
- Congenital Hyperinsulinism India Association (CHIA) https://www.hyperinsulinism-india.org/ (accessed on 10 November 2024)
Considering the above lacunae and to address the challenges in the management of such disorders, we have established the non-profit organisation, “Congenital Hyperinsulinism India Association”—CHI India Association (CHIA)—to dedicate our efforts in the field of Congenital Hyperinsulinism.
- Congenital Hyperinsulinism India (CHIA)—Vision
- ○
- To raise awareness of Congenital Hyperinsulinism as a rare genetic disorder among the public and healthcare professionals across India and contribute towards a better quality of life for the children and their families.
- Congenital Hyperinsulinism India Association (CHIA)—Mission
At CHIA, we dedicate our efforts in creating a one-stop solution for CHI in India. We plan to collaborate with individual clinicians as well as researchers and other organisations to unify efforts in the following:
- ○
- Offer prompt genetic testing for clinically and biochemically confirmed cases of CHI at the CAP (College of American Pathologists)- and NABL (National Accreditation Board for Testing and Calibration Laboratories)-accredited genetic centre in India to provide quick turn-around of results.
- ○
- Generate a national database and registry of patients for collaborative research, improving access to diagnostics and treatment.
- ○
- Formulate a parent support group and CHI-India Association consortium to raise awareness amongst families (with different vernacular regional languages) and healthcare professionals through national conferences and seminars, as well as government supported programmes.
- ○
- Design patient information leaflets (in different vernacular regional languages) to provide uniform and adequate care as well as ease of access to information.
- ○
- Identify and support potential centres of excellence in the country to expedite the diagnostic services in terms of genetic evaluation and imaging, as well as treatment modalities.
- ○
- Foster genomic collaborations with international organisations, as well as genetic centres (US and UK), to promote research collaborations in identifying and characterising novel functional gene mutations.
- ○
- Promote clinical trials for newer investigations and treatment modalities so children and families in India can obtain access to newer therapies.
2. Discussion
2.1. CHIA Patient Registry
An organised system to collect uniform data and evaluate specified outcomes in patients and families with CHI has been designed in accordance with the AHRQ guidelines [64]. The use of national registries for quality improvement purposes in rare diseases has been advocated in many countries and commonly serves the purpose of generating and formalising a data source, performing natural history studies, evaluating the association between clinical characteristics and health outcomes, understanding the phenotype–genotype correlations, establishing a network for future diagnostic and treatment studies, as well as supporting the patients to offer a more holistic care [65,66]. The CHIA registry consists of a series of questions, including demographic details, clinical history biochemical profile and treatment details, amongst others, to be filled by the patients/parents/guardians/consulting clinicians. The registry positions the patients and their families in a unique position to provide information related to their symptoms, functional status, quality of life and the overall burden of the disease to generate real-world data, and such patient reported outcomes could play an integral role in providing additional insight into the medical and day-today experience of living with a rare disease, as well as in clinical trial designs and regulatory decision making [67,68,69]. A GDPR (General Data Protection Regulations)-compliant database will also enable the electronic access of healthcare records in a mutually exclusive, ethical, and patient-friendly manner that could open more avenues for research in potential new treatments and improve quality of life.
2.2. CHIA—Healthcare Professionals’ Consortium
A CHI-India Association consortium has been developed that brings together a cadre of healthcare professionals like researchers, geneticists, clinicians, collaborators, as well as parents/patients to share relevant skills, experience and expertise in the field of CHI and thereby create a more user-friendly systems of care. The formally structured CHIA consortium is devised on the structural approach depending on the unique circumstances and problems pertaining to CHI in India. The consortium comprising its executive board functions to advance medical research through awareness campaigns outreach programmes and conferences along with community-wide needs assessment planning, enabling uniform high-quality data collection and analysis, data sharing, developing collateral resources and facilitating collaboration and information sharing across the network. More importantly, the consortium is envisaged to initiate cross-cutting collaborations between various national organisations of repute like Indian Academy of Pediatrics (IAP), National Neonatology Forum India (NNF India), ISPAE (Indian Society for Pediatric and Adolescent Endocrinology) as well as collaborate with global cooperation platforms like CHI International. This will enable us to develop national guidelines as well as identify centres of excellence for standardised CHI care in the country that will enable to create opportunities for advancing diagnostic and treatment options, including clinical trials for improved healthcare [70].
2.3. CHIA—Parent Support Group
A CHIA parent support group has been established to create a collective effort for awareness, policy issues, advocacy, research, and medical advancements, as well as emotional and psychological support to all patients and families with CHI. Similar parent support groups have yielded encouraging results, feeling empowered and a sense of belonging as they have been able to connect with each other and the healthcare professionals to provide emotional and financial support, as well as skills to deal with the day-to-day issues of raising children with rare disabilities [71,72].
2.4. CHIA—Information Leaflets
Considering the linguistic disparity and socio-cultural differences, information leaflets have been designed for patients as well as healthcare workers to create a high-quality source of good, accessible, and comprehensive information on the etio-pathogenesis, role of involved genes, and treatment options for CHI amongst others. With the advent of information and technology, patients have become increasingly engaged in their own healthcare, and these information leaflets serve in effectively communicating the information as well as in educating the stakeholders about the disease. It also provides a scope for creating a mutually beneficial partnership between patients, their families, and healthcare services that respects individual needs and values as well as demonstrates how shared decision making and patient empowerment enhance patient adherence to treatment [73,74,75]. Information leaflets that cater to the readability of individual patients, meet a global standard, and set up standardised guidelines to bring CHI on a uniform platform for all stakeholders have been designed (regional vernacular languages are in process). The way forward is to design and translate the leaflets for individual regional vernacular languages; disseminate these leaflets through awareness campaigns, social media platforms, and practice patient participation groups; and establish policies and protocols for patient care and management [76,77].
2.5. CHIA—Genetic Analysis and Genomic Collaborations
We have established communication channels with consultant paediatric endocrinologists and neonatologists along with clinical and molecular geneticists across India to coordinate investigations on next generation sequencing for the children and sanger sequencing for the parents. Information sheets about the CHI India Association, consent form, and a clinical proforma for generating a database are shared with these collaborators to conduct prompt testing and free-of-charge genetic analysis at the CAP (College of American Pathologists)- and NABL (National Accreditation Board for Testing and Calibration Laboratories)-accredited genetic centre “MDRF—Madras Diabetic Research Foundation” in India (www.mdrf.in & www.monogenicdiabetes.in (accessed on 10 November 2024)) to reduce the turn-around time of results. Moreover, with over one-third to one-half of the cases estimated to be of novel variants with unknown significance, we have collaborated with Genomic Medicine, University of Exeter Medical School and the Centre for Endocrinology, William Harvey Research Institute, Queen Mary University of London to identify and characterise the genes with novel mutations, as well as undertake functional work in future multi-centric genetic studies.
2.6. CHIA–Centres of Excellence
We aim to identify and collaborate with potential centres of excellence in the country to expedite the diagnostic services in terms of genetic evaluation and imaging, as well as treatment modalities, of rare diseases under the National Policy for Rare Diseases, 2021 [63].
3. Conclusions
Raising awareness regarding the burden of Congenital Hyperinsulinism in India to the lay population and medical practitioners is the need of the hour. Steps like CHIA may help to sensitise the health authorities and policy makers towards the need for an improvement in the allocation of health budgets, especially towards a rare disorder such as this. A national database for this rare disorder will be crucial in the empowerment of patient advocacy groups and proactive involvement of policy makers in the implementation of such programmes. While we understand that achieving such goals dedicated to CHI in India is hindered by several challenges, including the lack of awareness among the public and healthcare professionals, socio-economic disparities, competing healthcare priorities, and high treatment costs coupled with research and infrastructure gaps. At CHIA, we strongly urge the Government of India—including DST, ICMR, DBT, CSIR, and other agencies—along with philanthropists, corporate houses, pharmaceutical companies, private genetic laboratories, and foundations both in India and abroad, to join us in the fight against CHI in India and provide crucial support for such initiatives. We also aim to collaborate with national/international paediatric as well as neonatology organisations for furthering our goals. We offer memberships for the CHI-India Association consortium and CHI-India Association parent support group, and the links and registration page can be found on our website. Further information about the CHI-India Association can be found on www.hyperinsulinism-india.org.
Author Contributions
P.S. (Pratik Shah), V.M., V.R. and S.N. were responsible for the ideation and conceptualization of the article, as well as the availability of resources and supervision. J.B.C., V.R. and K.S. performed the literature search and data analysis for the article. J.B.C., P.S. (Praveen Singh), and S.T. were responsible for preparing the original draft of the article. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Ethics Committee of Pramukhswami Medical College, Bhaikaka University, Karamsad (R&D. No.: IEC/BU/137/Faculty/12/93/2022; Dated: 2 June 2022), registered with the National Ethics Committee Registry for Biomedical Health Research, Department of Health Research, Ministry of Health and Family Welfare, Government of India (File No.: EC/NEW/INST/2021/592).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original data presented in this study are openly available at www.hyperinsulinism-india.org (accessed on 5 January 2023).
Acknowledgments
The authors wish to acknowledge the contribution of the members of the CHIA consortium to the activities of the CHIA as well as to this article. We also wish to acknowledge the contributions of Surendra Kr. Choudhury, Suresh Kumar Khemka, and Sanjana Choudhury for their efforts as members of the parent support group for CHIA. We also acknowledge Congenital Hyperinsulinism International (CHI International), USA for collaborative work and Pratik Shah being part of the Collaborative Research Network for CHI International.
Conflicts of Interest
Author Sunil Tadepalli is employed by Labnetworx Health IT LLP. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AHRQ | Agency for Healthcare Research and Quality |
| CAP | College of American Pathologists |
| CHI | Congenital Hyperinsulinism |
| CHIA | Congenital Hyperinsulinism India Association |
| CSIR | Council of Scientific and Industrial Research |
| DBT | Department of Biotechnology |
| DST | Department of Science and Technology |
| GDP | Gross Domestic Product |
| GDPR | General Data Protection Regulations |
| IAP | Indian Association of Pediatrics |
| ICMR | Indian Council of Medical Research |
| ISPAE | Indian Society of Pediatric and Adolescent Endocrinologists |
| MDRF | Madras Diabetes Research Foundation |
| NABL | National Accreditation Board for Testing and Calibration Laboratories |
| NNF | National Neonatology Forum |
| NPRD | National Policy for Rare Diseases |
References
- Technical Group on Population Projections. Population projections for India and states 2011–2036. In Census of India 2011; National Commission on Population Ministry of Health & Family Welfare Nirman Bhawan: New Delhi, India, 2020. [Google Scholar]
- Majumder, P.P. The human genetic history of South Asia. Curr. Biol. 2010, 20, R184–R187. [Google Scholar] [CrossRef] [PubMed]
- Tamang, R.; Singh, L.; Thangaraj, K. Complex genetic origin of Indian populations and its implications. J. Biosci. 2012, 37, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Habib, I. A People’s History of India 1—Prehistory, 10th ed.; Tulika Books: New Delhi, India, 2015. [Google Scholar]
- Habib, I. A People’s History of India 2—The Indus Civilization, 9th ed.; Tulika Books: New Delhi, India, 2017. [Google Scholar]
- Yau, D.; Laver, T.W.; Dastamani, A.; Senniappan, S.; Houghton, J.A.L.; Shaikh, G.; Cheetham, T.; Mushtaq, T.; Kapoor, R.R.; Randell, T.; et al. Using referral rates for genetic testing to determine the incidence of a rare disease: The minimal incidence of congenital hyperinsulinism in the UK is 1 in 28,389. PLoS ONE 2020, 15, e0228417. [Google Scholar] [CrossRef]
- Juyal, G.; Mondal, M.; Luisi, P.; Laayouni, H.; Sood, A.; Midha, V.; Heutink, P.; Bertranpetit, J.; Thelma, B.K.; Casals, F. Population and genomic lessons from genetic analysis of two Indian populations. Hum. Genet. 2014, 133, 1273–1287. [Google Scholar] [CrossRef]
- Tripathi, M.; Tripathi, P.; Chauhan, U.K.; Herrera, R.J.; Agrawal, S. Alu polymorphic insertions reveal genetic structure of North Indian populations. Hum. Biol. 2008, 80, 483–499. [Google Scholar] [CrossRef]
- Bittles, A.H. Endogamy, consanguinity and community genetics. J. Genet. 2002, 81, 91–98. [Google Scholar] [CrossRef]
- Ministry of Finance. Economic Survey 2021–2022; Government of India: New Delhi, India, 2022.
- World Health Organization. World Health Organization Global Health Expenditure Database. Available online: http://apps.who.int/nha/database (accessed on 7 April 2023).
- Balarajan, Y.; Selvaraj, S.; Subramanian, S. Health care and equity in India. Lancet 2011, 377, 505–515. [Google Scholar] [CrossRef]
- Jindal, A.K. Universal health coverage: The way forward. Indian J. Public Health 2014, 58, 161–167. [Google Scholar] [CrossRef]
- Suresh, S.; Thangavel, G.; Sujatha, J.; Indrani, S. Methodological issues in setting up a surveillance system for birth defects in India. Natl. Med. J. India 2005, 18, 259–262. [Google Scholar]
- Phadke, S.R. Malformation syndromes in India. In Genetic Disorders in Indian Subcontinent; Kumar, D., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; pp. 155–166. [Google Scholar]
- Aggarwal, S.; Phadke, S.R. Medical genetics and genomic medicine in India: Current status and opportunities ahead. Mol. Genet. Genom. Med. 2015, 3, 160–171. [Google Scholar] [CrossRef]
- Thank you for sharing. Nat. Biotechnol. 2020, 38, 1005. [CrossRef]
- De Leon, D.D.; Stanley, C.A. Congenital Hypoglycemia Disorders: New Aspects of Etiology, Diagnosis, Treatment and Outcomes: Highlights of the Proceedings of the Congenital Hypoglycemia Disorders Symposium, Philadelphia April 2016. Pediatr. Diabetes 2016, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Raskin, J.; Arnoux, J.-B.; De Leon, D.D.; Weinzimer, S.A.; Hammer, M.; Kendall, D.M.; Thornton, P.S. Congenital hyperinsulinism in infancy and childhood: Challenges, unmet needs and the perspective of patients and families. Orphanet J. Rare Dis. 2022, 17, 61, Erratum in Orphanet J. Rare Dis. 2022, 17, 205. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ackermann, A.M.; Boodhansingh, K.E.; Bhatti, T.R.; Liu, C.; Schug, J.; Doliba, N.; Han, B.; Cosgrove, K.E.; Banerjee, I.; et al. Functional and Metabolomic Consequences of KATP Channel Inactivation in Human Islets. Diabetes 2017, 66, 1901–1913. [Google Scholar] [CrossRef]
- UNICEF India. Newborn and Child Health. Available online: https://www.unicef.org/india/what-we-do/newborn-and-child-health (accessed on 16 July 2023).
- Muukkonen, L.; Männistö, J.; Jääskeläinen, J.; Hannonen, R.; Huopio, H. The effect of hypoglycaemia on neurocognitive outcome in children and adolescents with transient or persistent congenital hyperinsulinism. Dev. Med. Child Neurol. 2019, 61, 451–457. [Google Scholar] [CrossRef]
- Shah, P.; Rahman, S.A.; Demirbilek, H.; Güemes, M.; Hussain, K. Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol. 2017, 5, 729–742. [Google Scholar] [CrossRef]
- Sharma, R.; Roy, K.; Satapathy, A.K.; Kumar, A.; Nanda, P.M.; Damle, N.; Houghton, J.A.L.; Flanagan, S.E.; Radha, V.; Mohan, V.; et al. Molecular Characterization and Management of Congenital Hyperinsulinism: A Tertiary Centre Experience. Indian Pediatr. 2022, 59, 105–109. [Google Scholar] [CrossRef]
- Nestorowicz, A.; Inagaki, N.; Gonoi, T.; Schoor, K.P.; Wilson, B.A.; Glaser, B.; Landau, H.; Stanley, C.A.; Thornton, P.S.; Seino, S.; et al. A nonsense mutation in the inward rectifier potassium channel gene, Kir6.2, is associated with familial hyperinsulinism. Diabetes 1997, 46, 1743–1748. [Google Scholar] [CrossRef]
- Jahnavi, S.; Poovazhagi, V.; Kanthimathi, S.; Balamurugan, K.; Bodhini, D.; Yadav, J.; Jain, V.; Khadgawat, R.; Sikdar, M.; Bhavatharini, A.; et al. NovelABCC8(SUR1) Gene mutations in Asian Indian children with Congenital Hyperinsulinemic Hypoglycemia. Ann. Hum. Genet. 2014, 78, 311–319. [Google Scholar] [CrossRef]
- Thomas, P.M.; Cote, G.J.; Wohllk, N.; Haddad, B.; Mathew, P.M.; Rabl, W.; Aguilar-Bryan, L.; Gagel, R.F.; Bryan, J. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 1995, 268, 426–429. [Google Scholar] [CrossRef]
- Babenko, A.P.; Polak, M.; Cavé, H.; Busiah, K.; Czernichow, P.; Scharfmann, R.; Bryan, J.; Aguilar-Bryan, L.; Vaxillaire, M.; Froguel, P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 2006, 355, 456–466. [Google Scholar] [CrossRef]
- Shah, I.A.; Rashid, R.; Bhat, A.; Rashid, H.; Bashir, R.; Asrar, M.M.; Wani, I.A.; Charoo, B.A.; Radha, V.; Mohan, V.; et al. A novel mutation in the KCNJ11 gene (p.Val36Glu), predisposes to congenital hyperinsulinemia. Gene 2023, 878, 147576. [Google Scholar] [CrossRef]
- Roy, K.; Satapathy, A.K.; Houhton, J.A.L.; Flanagan, S.E.; Radha, V.; Mohan, V.; Sharma, R.; Jain, V. Congenital Hyperinsulinemic Hypoglycemia and Hyperammonemia due to Pathogenic Variants in GLUD1. Indian J. Pediatr. 2019, 86, 1051–1053. [Google Scholar] [CrossRef]
- Li, C.; Chen, P.; Palladino, A.; Narayan, S.; Russell, L.K.; Sayed, S.; Xiong, G.; Chen, J.; Stokes, D.; Butt, Y.M.; et al. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J. Biol. Chem. 2010, 285, 31806–31818. [Google Scholar] [CrossRef] [PubMed]
- Glaser, B.; Kesavan, P.; Heyman, M.; Davis, E.; Cuesta, A.; Buchs, A.; Stanley, C.A.; Thornton, P.S.; Permutt, M.A.; Matschinsky, F.M.; et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N. Engl. J. Med. 1998, 338, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.J.; Kapoor, R.R.; Eaton, S.; Chadefaux, B.; Akcay, T.; Simsek, E.; E Flanagan, S.; Ellard, S.; Hussain, K. Leucine-sensitive hyperinsulinaemic hypoglycaemia in patients with loss of function mutations in 3-Hydroxyacyl-CoA Dehydrogenase. Orphanet J. Rare Dis. 2012, 7, 25. [Google Scholar] [CrossRef]
- Molven, A.; Matre, G.E.; Duran, M.; Wanders, R.J.; Rishaug, U.; Njølstad, P.R.; Jellum, E.; Søvik, O. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes 2004, 53, 221–227. [Google Scholar] [CrossRef]
- Clayton, P.T.; Eaton, S.; Aynsley-Green, A.; Edginton, M.; Hussain, K.; Krywawych, S.; Datta, V.; Malingré, H.E.; Berger, R.; Berg, I.E.V.D. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of β-oxidation in insulin secretion. J. Clin. Investig. 2001, 108, 457–465. [Google Scholar] [CrossRef]
- Verdecchia, F.; Akcan, N.; Dastamani, A.; Morgan, K.; Semple, R.K.; Shah, P. Unusual glycemic presentations in a child with a novel heterozygous intragenic INSR deletion. Horm. Res. Paediatr. 2020, 93, 396–401. [Google Scholar] [CrossRef]
- Sethi, A.; Foulds, N.; Ehtisham, S.; Ahmed, S.H.; Houghton, J.; Colclough, K.; Didi, M.; Flanagan, S.E.; Senniappan, S. Heterozygous Insulin Receptor (INSR) Mutation Associated with Neonatal Hyperinsulinemic Hypoglycaemia and Familial Diabetes Mellitus: Case Series. J. Clin. Res. Pediatr. Endocrinol. 2020, 12, 420–426. [Google Scholar] [CrossRef]
- Ferrara, C.T.; Boodhansingh, K.E.; Paradies, E.; Giuseppe, F.; Steinkrauss, L.J.; Topor, L.S.; Quintos, J.B.; Ganguly, A.; De Leon, D.D.; Palmieri, F.; et al. Novel Hypoglycemia Phenotype in Congenital Hyperinsulinism Due to Dominant Mutations of Uncoupling Protein 2. J. Clin. Endocrinol. Metab. 2017, 102, 942–949, Erratum in J. Clin. Endocrinol. Metab. 2018, 103, 2076. [Google Scholar] [CrossRef]
- González-Barroso, M.M.; Giurgea, I.; Bouillaud, F.; Anedda, A.; Bellanné-Chantelot, C.; Hubert, L.; de Keyzer, Y.; de Lonlay, P.; Ricquier, D. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS ONE 2008, 3, e3850. [Google Scholar] [CrossRef]
- Tung, J.Y.-L.; Boodhansingh, K.; Stanley, C.A.; De León, D.D. Clinical heterogeneity of hyperinsulinism due to HNF1A and HNF4A mutations. Pediatr. Diabetes 2018, 19, 910–916. [Google Scholar] [CrossRef]
- McGlacken-Byrne, S.M.; Mohammad, J.K.; Conlon, N.; Gubaeva, D.; Siersbæk, J.; Schou, A.J.; Demirbilek, H.; Dastamani, A.; Houghton, J.A.L.; Brusgaard, K.; et al. Clinical and genetic heterogeneity of HNF4A/HNF1A mutations in a multicentre paediatric cohort with hyperinsulinaemic hypoglycaemia. Eur. J. Endocrinol. 2022, 186, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Dusatkova, P.; Pruhova, S.; Sumnik, Z.; Kolouskova, S.; Obermannova, B.; Cinek, O.; Lebl, J. HNF1A mutation presenting with fetal macrosomia and hypoglycemia in childhood prior to onset of overt diabetes. J. Pediatr. Endocrinol. Metab. 2011, 24, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Pingul, M.M.; Hughes, N.; Wu, A.; Stanley, C.A.; Gruppuso, P.A. Hepatocyte nuclear factor 4α gene mutation associated with familial neonatal hyperinsulinism and maturity-onset diabetes of the young. J. Pediatr. 2011, 158, 852–854. [Google Scholar] [CrossRef]
- E Flanagan, S.; Kapoor, R.R.; Mali, G.; Cody, D.; Murphy, N.; Schwahn, B.; Siahanidou, T.; Banerjee, I.; Akcay, T.; Rubio-Cabezas, O.; et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur. J. Endocrinol. 2010, 162, 987–992. [Google Scholar] [CrossRef]
- Pearson, E.R.; Boj, S.F.; Steele, A.M.; Barrett, T.; Stals, K.; Shield, J.P.; Ellard, S.; Ferrer, J.; Hattersley, A.T. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007, 4, e118. [Google Scholar] [CrossRef]
- Pinney, S.E.; Ganapathy, K.; Bradfield, J.; Stokes, D.; Sasson, A.; Mackiewicz, K.; Boodhansingh, K.; Hughes, N.; Becker, S.; Givler, S.; et al. Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm. Res. Paediatr. 2013, 80, 18–27. [Google Scholar] [CrossRef]
- Torekov, S.S.; Iepsen, E.; Christiansen, M.; Linneberg, A.; Pedersen, O.; Holst, J.J.; Kanters, J.K.; Hansen, T. KCNQ1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes 2014, 63, 1315–1325. [Google Scholar] [CrossRef]
- Kostopoulou, E.; Dastamani, A.; Güemes, M.; Clement, E.; Caiulo, S.; Shanmugananda, P.; Dattani, M.; Gilbert, C.; A Hurst, J.; Shah, P. Syndromic Forms of Hyperinsulinaemic Hypoglycaemia—A 15-year follow-up Study. Clin. Endocrinol. 2021, 94, 399–412. [Google Scholar] [CrossRef]
- Giri, D.; Vignola, M.L.; Gualtieri, A.; Scagliotti, V.; McNamara, P.; Peak, M.; Didi, M.; Gaston-Massuet, C.; Senniappan, S. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Hum. Mol. Genet. 2017, 26, 4315–4326. [Google Scholar] [CrossRef] [PubMed]
- Ocansey, S.; Pullen, D.; Atkinson, P.; Clarke, A.; Hadonou, M.; Crosby, C.; Short, J.; Lloyd, I.C.; Smedley, D.; Assunta, A.; et al. Biallelic DNAJC3 variants in a neuroendocrine developmental disorder with insulin dysregulation. Clin. Dysmorphol. 2022, 31, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Gϋemes, M.; Rahman, S.A.; Kapoor, R.R.; Flanagan, S.; Houghton, J.A.L.; Misra, S.; Oliver, N.; Dattani, M.T.; Shah, P. Hyperinsulinemic hypoglycemia in children and adolescents: Recent advances in understanding of pathophysiology and management. Rev. Endocr. Metab. Disord. 2020, 21, 577–597. [Google Scholar] [CrossRef] [PubMed]
- Hewat, T.I.; Johnson, M.B.; Flanagan, S.E. Congenital Hyperinsulinism: Current Laboratory-Based Approaches to the Genetic Diagnosis of a Heterogeneous Disease. Front. Endocrinol. 2022, 13, 873254. [Google Scholar] [CrossRef]
- Galcheva, S.; Demirbilek, H.; Al-Khawaga, S.; Hussain, K. The Genetic and Molecular Mechanisms of Congenital Hyperinsulinism. Front. Endocrinol. 2019, 10, 111. [Google Scholar] [CrossRef]
- Kapoor, R.R.; Flanagan, S.E.; Arya, V.B.; Hamilton-Shield, J.P.; Ellard, S.; Hussain, K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur. J. Endocrinol. 2013, 168, 557–564. [Google Scholar] [CrossRef]
- Snider, K.E.; Becker, S.; Boyajian, L.; Shyng, S.-L.; MacMullen, C.; Hughes, N.; Ganapathy, K.; Bhatti, T.; Stanley, C.A.; Ganguly, A. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J. Clin. Endocrinol. Metab. 2013, 98, E355–E363. [Google Scholar] [CrossRef]
- Aftab, S.; Gubaeva, D.; Houghton, J.A.L.; Dastamani, A.; Sotiridou, E.; Gilbert, C.; Flanagan, S.E.; Tiulpakov, A.; Melikyan, M.; Shah, P. Spectrum of neuro-developmental disorders in children with congenital hyperinsulinism due to activating mutations in GLUD1. Endocr. Connect. 2023, 12, e220008. [Google Scholar] [CrossRef]
- Banerjee, I.; Salomon-Estebanez, M.; Shah, P.; Nicholson, J.; Cosgrove, K.E.; Dunne, M.J. Therapies and outcomes of congenital hy-perinsulinism-induced hypoglycaemia. Diabet. Med. 2019, 36, 9–21. [Google Scholar]
- Roženková, K.; Güemes, M.; Shah, P.; Hussain, K. The Diagnosis and Management of Hyperinsulinaemic Hypoglycaemia. J. Clin. Res. Pediatr. Endocrinol. 2015, 7, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Welters, A.; Lerch, C.; Kummer, S.; Marquard, J.; Salgin, B.; Mayatepek, E.; Meissner, T. Long-term medical treatment in congenital hyperinsulinism: A descriptive analysis in a large cohort of patients from different clinical centers. Orphanet J. Rare Dis. 2015, 10, 150. [Google Scholar] [CrossRef]
- Fékété, C.; de Lonlay, P.; Jaubert, F.; Rahier, J.; Brunelle, F.; Saudubray, J. The surgical management of congenital hyperinsulinemic hypoglycemia in infancy. J. Pediatr. Surg. 2004, 39, 267–269. [Google Scholar] [CrossRef]
- Welters, A.; Meissner, T.; Grulich-Henn, J.; Fröhlich-Reiterer, E.; Warncke, K.; Mohnike, K.; Blankenstein, O.; Menzel, U.; Datz, N.; Bollow, E.; et al. Characterization of diabetes following pancreatic surgery in patients with congenital hyperinsulinism. Orphanet J. Rare Dis. 2018, 13, 230. [Google Scholar] [CrossRef] [PubMed]
- Rajasimha, H.K.; Shirol, P.B.; Ramamoorthy, P.; Hegde, M.; Barde, S.; Chandru, V.; Ravinandan, M.E.; Ramchandran, R.; Haldar, K.; Lin, J.C.; et al. Organization for rare diseases India (ORDI)—Addressing the challenges and opportunities for the Indian rare diseases’ community. Genet. Res. 2014, 96, e009. [Google Scholar] [CrossRef]
- Ministry of Health and Family Welfare. National Policy for Rare Diseases, 2021; Government of India: New Delhi, India, 2021.
- Gliklich, R.E.; Dreyer, N.A.; Leavy, M.B. (Eds.) Registries for Evaluating Patient Outcomes: A User’s Guide, 4th ed.; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2020.
- Richesson, R.; Vehik, K. Patient registries: Utility, validity and inference. Adv. Exp. Med. Biol. 2010, 686, 87–104. [Google Scholar] [CrossRef]
- Boulanger, V.; Schlemmer, M.; Rossov, S.; Seebald, A.; Gavin, P. Establishing Patient Registries for Rare Diseases: Rationale and Challenges. Pharm. Med. 2020, 34, 185–190. [Google Scholar] [CrossRef]
- der Weide, M.C.J.-V.; Gaasterland, C.M.W.; Roes, K.C.B.; Pontes, C.; Vives, R.; Sancho, A.; Nikolakopoulos, S.; Vermeulen, E.; van der Lee, J.H. Rare disease registries: Potential applications towards impact on development of new drug treatments. Orphanet J. Rare Dis. 2018, 13, 154. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research; Center for Biologics Evaluation and Research; Oncology Center of Excellence. Real-World Data: Assessing Registries to Support Regulatory Decision-Making for Drug and Biological Products Guidance for Industry; U.S. Food and Drug Administration: Rockville, MD, USA, 2021.
- Wu, J.; Wang, C.; Toh, S.; Pisa, F.E.; Bauer, L. Use of real-world evidence in regulatory decisions for rare diseases in the United States—Current status and future directions. Pharmacoepidemiol. Drug Saf. 2020, 29, 1213–1218. [Google Scholar] [CrossRef]
- Caplan, P.A.; Lefkowitz, B.; Spector, L. Health care consortia: A mechanism for increasing access for the medically indigent. Henry Ford. Hosp. Med. J. 1992, 40, 50–55. [Google Scholar]
- Zeutenhorst, R. Parent Support Groups and Well-Being: Investigating the Benefits of Parent Support Groups for Families of Children with Special Needs. Master’s Thesis, DORDT University, Sioux Center, IA, USA, 2017. [Google Scholar]
- Law, M.; King, S.; Stewart, D.; King, G. The perceived effects of parent-led support groups for parents of children with disabilities. Phys. Occup. Ther. Pediatr. 2001, 21, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Poplas-Susič, T.; Klemenc-Ketis, Z.; Kersnik, J. Usefulness of the patient information leaflet (PIL) and information on medicines from professionals: A patients’ view. A qualitative study. Slov. Med. J. 2014, 83, 368–375. [Google Scholar]
- Nathan, J.P.; Zerilli, T.; Cicero, L.A.; Rosenberg, J.M. Patients′ use and perception of medication information leaflets. Ann. Pharmacother. 2007, 41, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Gupta, U.; Sharma, S.; Sheth, P.D.; Jha, J.; Chaudhury, R.R. Improving medicine usage through patient information leaflets in India. Trop. Dr. 2005, 35, 164–166. [Google Scholar] [CrossRef]
- Vinker, S.; Eliyahu, V.; Yaphe, J. The effect of drug information leaflets on patient behavior. Isr. Med. Assoc. J. 2007, 9, 383–386. [Google Scholar]
- Schwappach, D.L.; Mülders, V.; Simic, D.; Wilm, S.; Thürmann, P.A. Is less more? Patients’ preferences for drug information leaflets. Pharmacoepidemiol. Drug Saf. 2011, 20, 987–995. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).