Neptunium(V) and Uranium(VI) Reactions at the Magnetite (111) Surface

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Grazing Incidence X-ray Absorption Spectroscopy (GI-XAS)
2.2. X-ray Photoemission Spectroscopy (XPS)
3. Results and Discussion
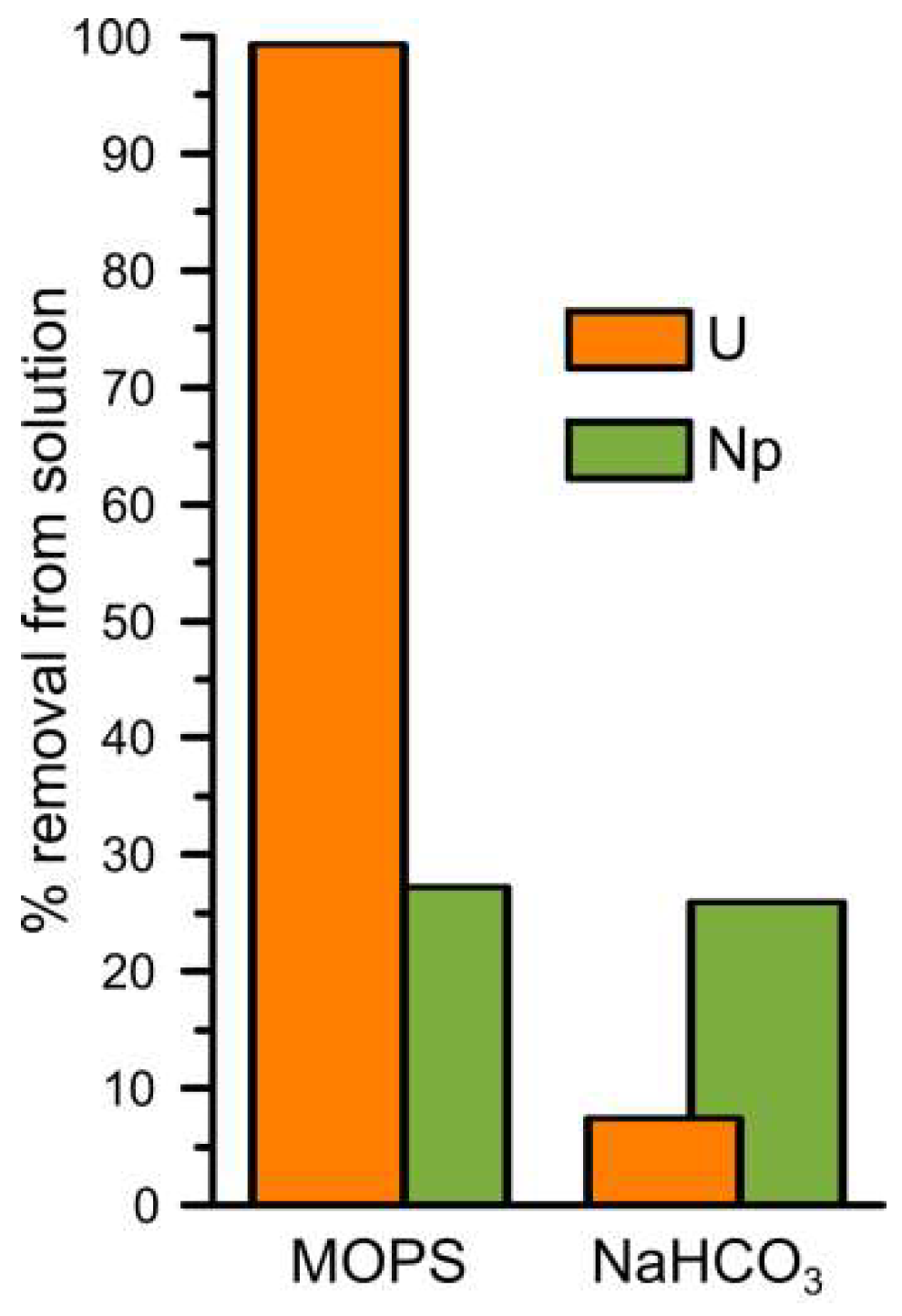
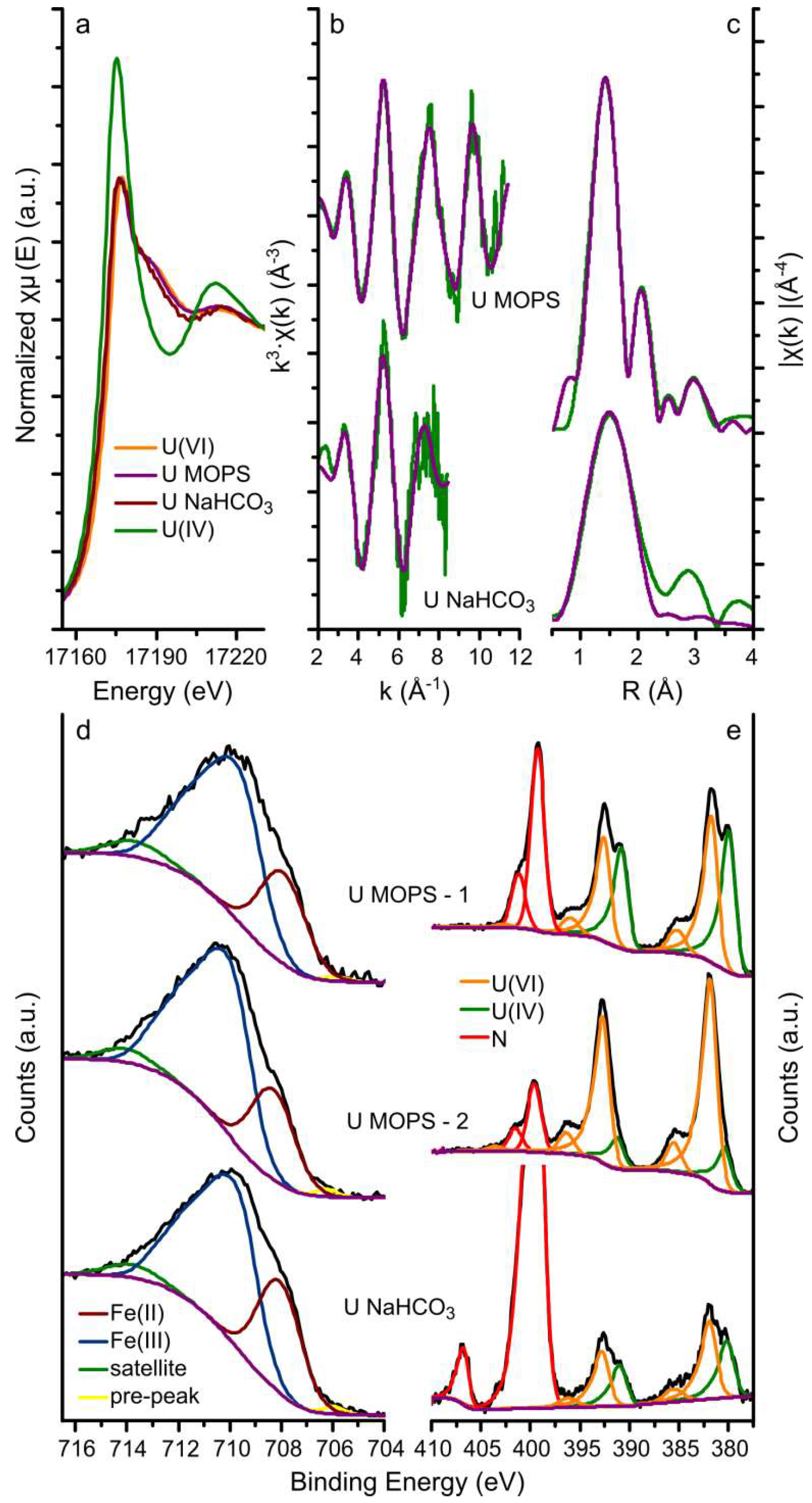
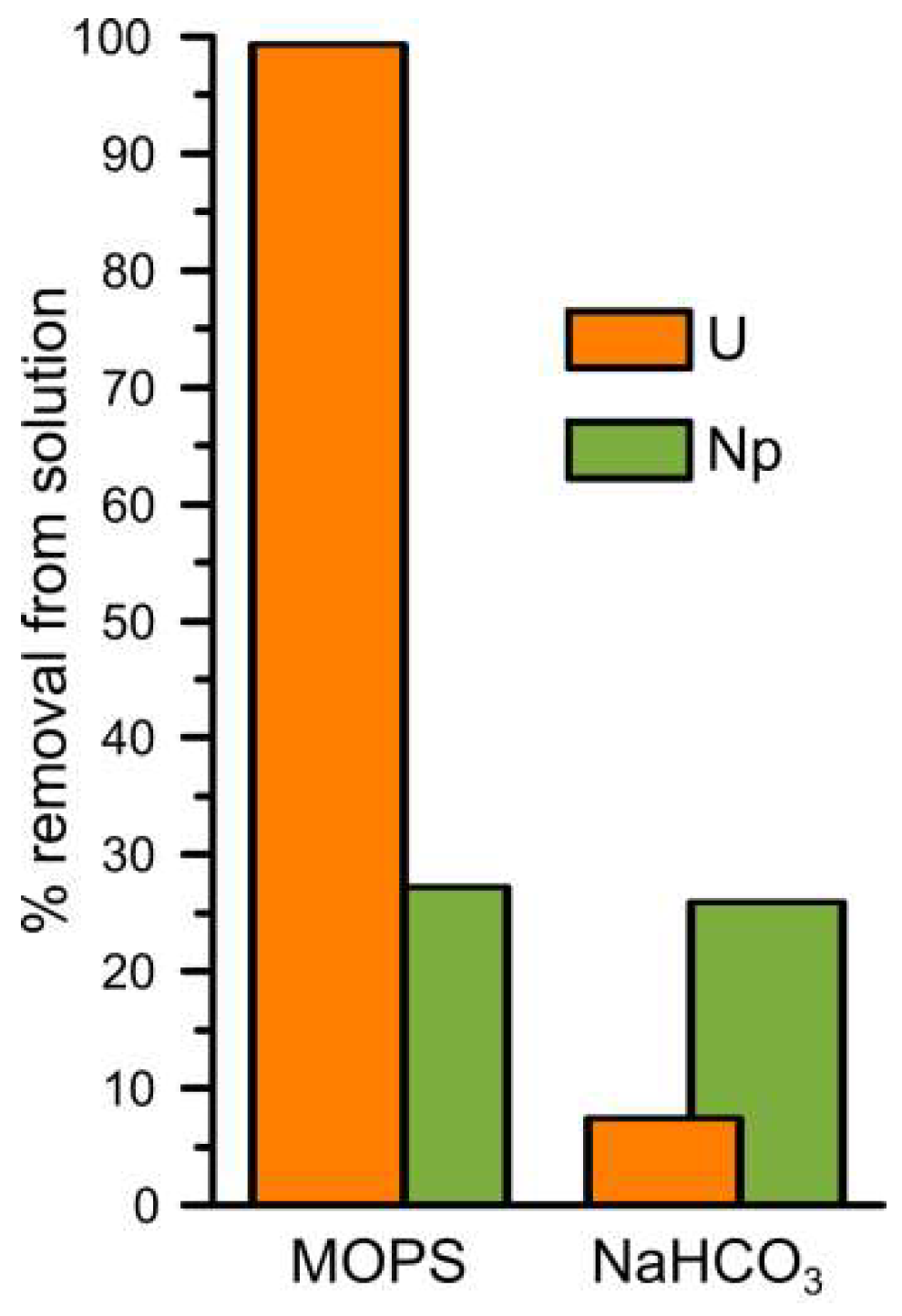
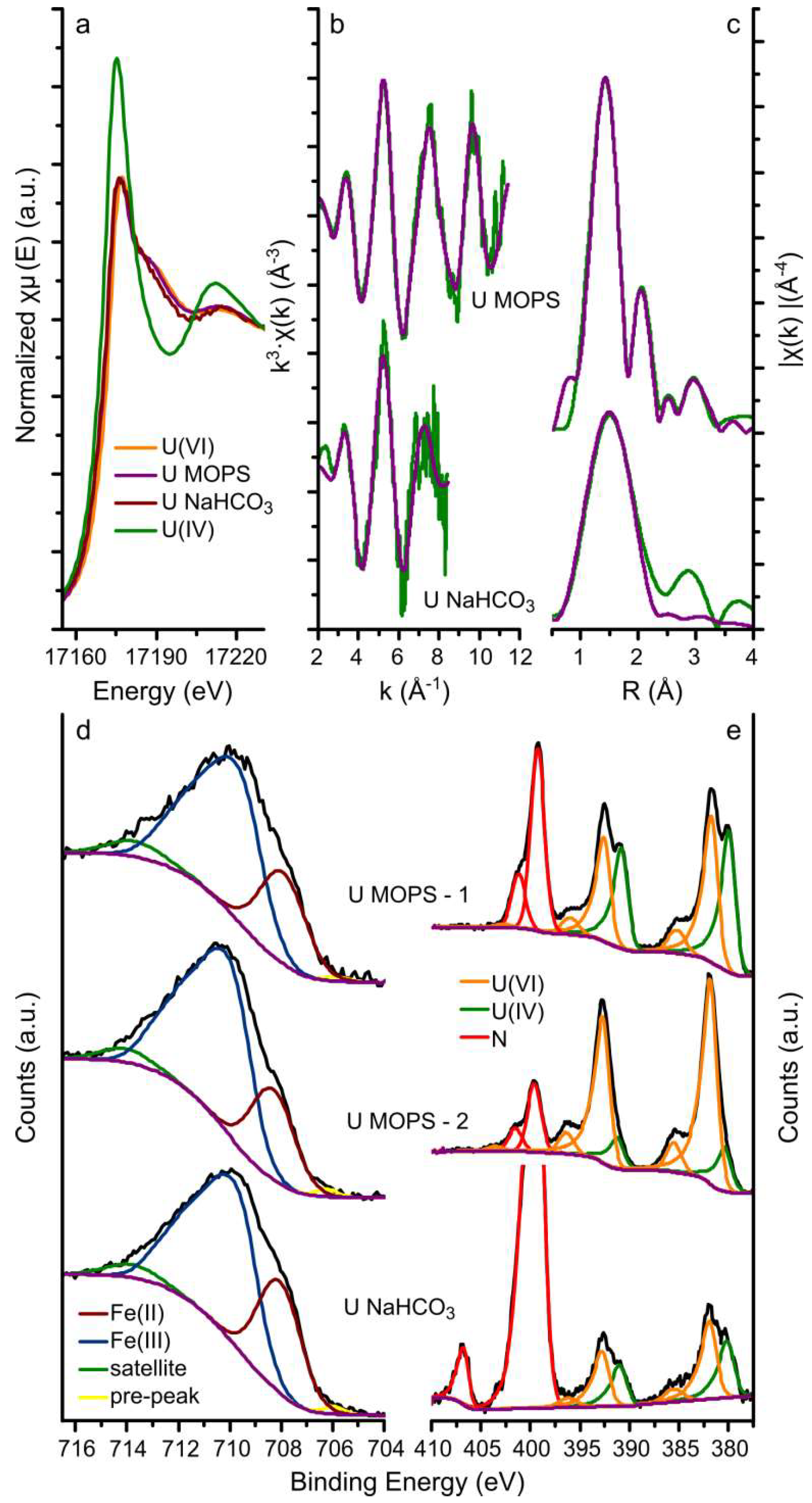
3.1. Uranium Interaction with Magnetite
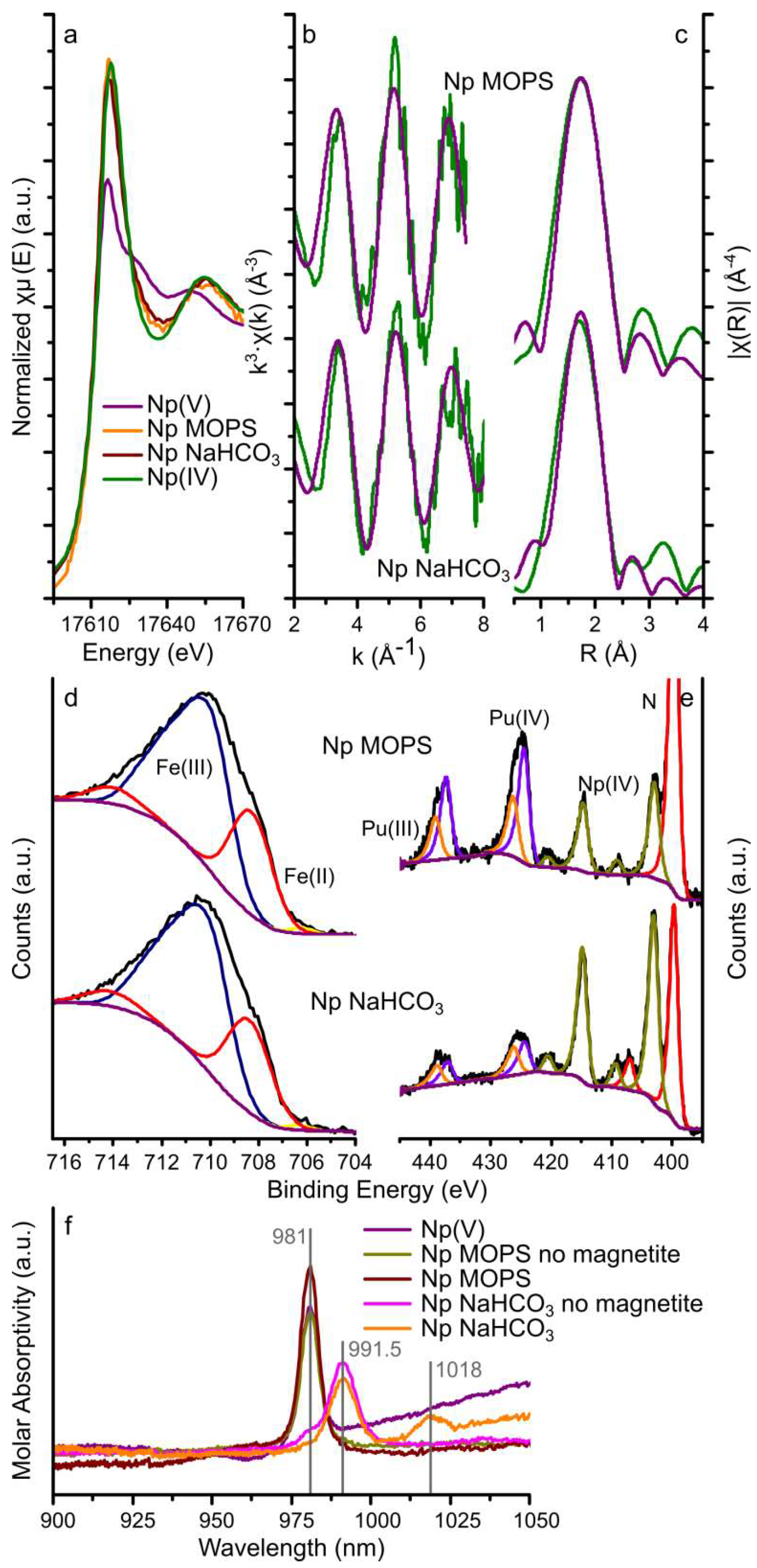
3.2. Neptunium (and Plutonium) Interactions with Magnetite
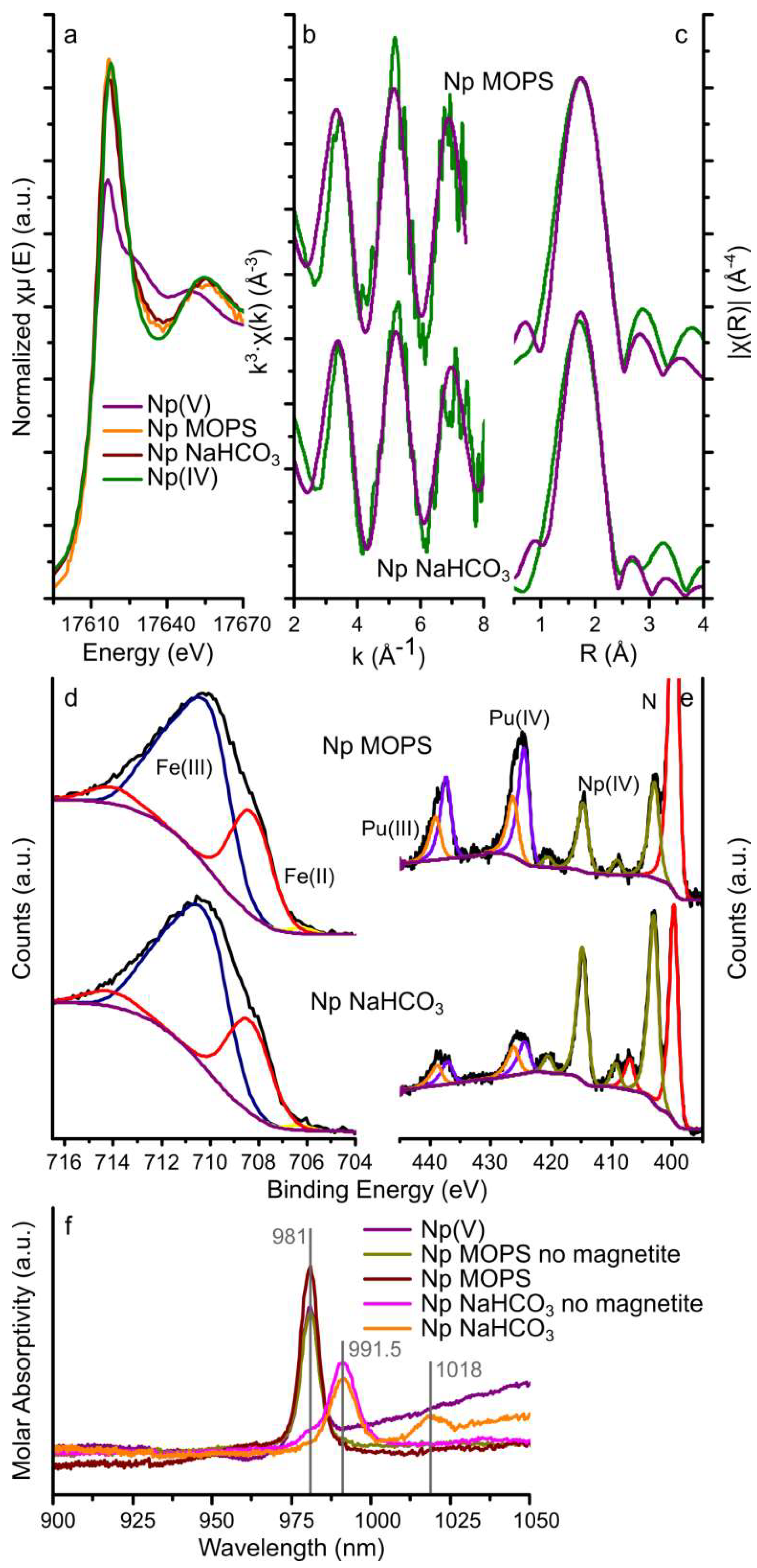
3.2.1. Neptunium
3.2.2. Plutonium
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morris, K.; Law, G.T.W.; Bryan, N.D. Geodisposal of Higher Activity Wastes. In Nuclear Power and the Environment; The Royal Society of Chemistry: London, UK, 2011; pp. 129–151. [Google Scholar]
- Cantrell, K.J.; Um, W.; Williams, B.D.; Bowden, M.E.; Gartman, B.; Lukens, W.W.; Buck, E.C.; Mausolf, E.J. Chemical stabilization of Hanford tank residual waste. J. Nucl. Mater. 2014, 446, 246–256. [Google Scholar] [CrossRef]
- Cendón, D.I.; Hughes, C.E.; Harrison, J.J.; Hankin, S.I.; Johansen, M.P.; Payne, T.E.; Wong, H.; Rowling, B.; Vine, M.; Wilsher, K.; et al. Identification of sources and processes in a low-level radioactive waste site adjacent to landfills: Groundwater hydrogeochemistry and isotopes. Aust. J. Earth Sci. 2015, 62, 123–141. [Google Scholar] [CrossRef]
- Payne, T.E.; Harrison, J.J.; Hughes, C.E.; Johansen, M.P.; Thiruvoth, S.; Wilsher, K.L.; Cendón, D.I.; Hankin, S.I.; Rowling, B.; Zawadzki, A. Trench ‘Bathtubbing’ and surface plutonium contamination at a legacy radioactive waste site. Environ. Sci. Technol. 2013, 47, 13284–13293. [Google Scholar] [CrossRef]
- Anderson, R.T.; Vrionis, H.A.; Ortiz-Bernad, I.; Resch, C.T.; Long, P.E.; Dayvault, R.; Karp, K.; Marutzky, S.; Metzler, D.R.; Peacock, A.; et al. Stimulating the in situ activity of Geobacter species to remove uranium from the groundwater of a uranium-contaminated aquifer. Appl. Environ. Microbiol. 2003, 69, 5884–5891. [Google Scholar] [CrossRef]
- Suzuki, Y.; Kelly, S.D.; Kemner, K.M.; Banfield, J.F. Microbial populations stimulated for hexavalent uranium reduction in uranium mine sediment. Appl. Environ. Microbiol. 2003, 69, 1337–1346. [Google Scholar] [CrossRef]
- Law, G.T.W.; Geissler, A.; Burke, I.T.; Livens, F.R.; Lloyd, J.R.; McBeth, J.M.; Morris, K. Uranium redox cycling in sediment and biomineral systems. Geomicrobiol. J. 2011, 28, 497–506. [Google Scholar] [CrossRef]
- Marsdem, O.J.; Abrahamsen, L.; Brayan, N.D.; Day Philip, J.; Fifield, K.; Gent, C.; Goodal, P.S.; Morris, K.; Livens, F.R. Transport and accumulation of actinide elements in the near-shore environment: Field and modelling studies. Sedimentology 2006, 53, 237–248. [Google Scholar] [CrossRef]
- Duro, L.; Domènech, C.; Grivé, M.; Roman-Ross, G.; Bruno, J.; Källström, K. Assessment of the evolution of the redox conditions in a low and intermediate level nuclear waste repository (SFR1, Sweden). Appl. Geochem. 2014, 49, 192–205. [Google Scholar] [CrossRef]
- Bots, P.; Shaw, S.; Law, G.T.W.; Marshall, T.A.; Mosselmans, J.F.W.; Morris, K. Controls on the Fate and Speciation of Np(V) during Iron (Oxyhydr)oxide Crystallization. Environ. Sci. Technol. 2016, 50, 3382–3390. [Google Scholar] [CrossRef]
- Marshall, T.A.; Morris, K.; Law, G.T.W.; Livens, F.R.; Mosselmans, J.F.W.; Bots, P.; Shaw, S. Incorporation of uranium into hematite during crystallization from ferrihydrite. Environ. Sci. Technol. 2014, 48, 3724–3731. [Google Scholar] [CrossRef]
- Marshall, T.A.; Morris, K.; Law, G.T.W.; Mosselmans, F.W.; Bots, P.; Roberts, H.; Shaw, S. Uranium fate during crystallization of magnetite from ferrihydrite in conditions relevant to the disposal of radioactive waste. Miner. Mag. 2015, 79, 1265–1274. [Google Scholar] [CrossRef]
- Powell, B.A.; Fjeld, R.A.; Kaplan, D.I.; Coates, J.T.; Serkiz, S.M. Pu(V)O2+ Adsorption and Reduction by Synthetic Magnetite (Fe3O4). Environ. Sci. Technol. 2004, 38, 6016–6024. [Google Scholar] [CrossRef]
- Tinnacher, R.M.; Zavarin, M.; Powell, B.A.; Kersting, A.B. Kinetics of neptunium(V) sorption and desorption on goethite: An experimental and modeling study. Geochim. Cosmochim. Acta 2011, 75, 6584–6599. [Google Scholar] [CrossRef]
- Wong, J.C.; Zavarin, M.; Begg, J.D.; Kersting, A.B.; Powell, B.A. Effect of equilibration time on Pu desorption from goethite. Radiochim. Acta 2015, 103, 695–705. [Google Scholar] [CrossRef]
- Wylie, E.M.; Olive, D.T.; Powell, B.A. Effects of Titanium Doping in Titanomagnetite on Neptunium Sorption and Speciation. Environ. Sci. Technol. 2016, 50, 1853–1858. [Google Scholar] [CrossRef]
- Kirsch, R.; Fellhauer, D.; Altmaier, M.; Neck, V.; Rossberg, A.; Fanghänel, T.; Charlet, L.; Scheinost, A.C. Oxidation state and local structure of plutonium reacted with magnetite, mackinawite, and chukanovite. Environ. Sci. Technol. 2011, 45, 7267–7274. [Google Scholar] [CrossRef]
- Pidchenko, I.; Heberling, F.; Kvashnina, K.O.; Finck, N.; Schild, D.; Bohnert, E.; Schäfer, T.; Rothe, J.; Geckeis, H.; Vitova, T. Aqueous U(VI) interaction with magnetite nanoparticles in a mixed flow reactor system: HR-XANES study. J. Phys. Conf. Ser. 2016, 712, 012086. [Google Scholar] [CrossRef]
- Li, D.; Kaplan, D.I. Sorption coefficients and molecular mechanisms of Pu, U, Np, Am and Tc to Fe (hydr)oxides: A review. J. Hazard. Mater. 2012, 243, 1–18. [Google Scholar] [CrossRef]
- Waite, T.D.; Davis, J.A.; Payne, T.E.; Waychunas, G.A.; Xu, N. Uranium(VI) adsorption to ferrihydrite: Application of a surface complexation model. Geochim. Cosmochim. Acta 1994, 58, 5465–5478. [Google Scholar] [CrossRef]
- Roberts, H.E.; Morris, K.; Law, G.T.W.; Mosselmans, J.F.W.; Bots, P.; Kvashnina, K.; Shaw, S. Uranium(V) Incorporation Mechanisms and Stability in Fe(II)/Fe(III) (oxyhydr)Oxides. Environ. Sci. Technol. Lett. 2017, 4, 421–426. [Google Scholar] [CrossRef]
- Guillaumont, R.; Fanghänel, T.; Neck, V.; Guger, J.; Palmer, D.A.; Grenthe, I.; Rand, M.H. Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium; OECD Nuclear Energy Agency, Data Bank: Issy-les-Moulineaux, France, 2003. [Google Scholar]
- Latta, D.E.; Gorski, C.A.; Boyanov, M.I.; O’Loughlin, E.J.; Kemner, K.M.; Scherer, M.M. Influence of magnetite stoichiometry on U VI reduction. Environ. Sci. Technol. 2012, 46, 778–786. [Google Scholar] [CrossRef]
- Latta, D.E.; Mishra, B.; Cook, R.E.; Kemner, K.M.; Boyanov, M.I. Stable U(IV) Complexes Form at High-Affinity Mineral Surface Sites. Environ. Sci. Technol. 2014. [Google Scholar] [CrossRef]
- Latta, D.E.; Boyanov, M.I.; Kemner, K.M.; O’Loughlin, E.J.; Scherer, M.M. Abiotic reduction of uranium by Fe(II) in soil. Appl. Geochem. 2012, 27, 1512–1524. [Google Scholar] [CrossRef]
- Scott, T.B.; Allen, G.C.; Heard, P.J.; Randell, M.G. Reduction of U(VI) to U(IV) on the surface of magnetite. Geochim. Cosmochim. Acta 2005, 69, 5639–5646. [Google Scholar] [CrossRef]
- Singer, D.M.; Chatman, S.M.; Ilton, E.S.; Rosso, K.M.; Banfield, J.F.; Waychunas, G.A. Identification of simultaneous U(VI) sorption complexes and U(IV) nanoprecipitates on the magnetite (111) surface. Environ. Sci. Technol. 2012, 46, 3811–3820. [Google Scholar] [CrossRef]
- Nakata, K.; Nagasaki, S.; Tanaka, S.; Sakamoto, Y.; Tanaka, T.; Ogawa, H. Reduction rate of neptunium(V) in heterogeneous solution with magnetite. Radiochim. Acta 2004, 92, 145–149. [Google Scholar] [CrossRef]
- Tang, J.; Myers, M.; Bosnick, K.A.; Brus, L.E. Magnetite Fe3O4 Nanocrystals: Spectroscopic Observation of Aqueous Oxidation Kinetics. J. Phys. Chem. B 2003, 107, 7501–7506. [Google Scholar] [CrossRef]
- White, A.F.; Peterson, M.L.; Hochella, M.F. Electrochemistry and dissolution kinetics of magnetite and ilmenite. Geochim. Cosmochim. Acta 1994, 58, 1859–1875. [Google Scholar] [CrossRef]
- Huber, F.; Schild, D.; Vitova, T.; Rothe, J.; Kirsch, R.; Schäfer, T. U(VI) removal kinetics in presence of synthetic magnetite nanoparticles. Geochim. Cosmochim. Acta 2012, 96, 154–173. [Google Scholar] [CrossRef]
- Ilton, E.S.; Boily, J.F.S.; Buck, E.C.; Skomurski, F.N.; Rosso, K.M.; Cahill, C.L.; Bargar, J.R.; Felmy, A.R. Influence of dynamical conditions on the reduction of UVI at the magnetite-solution interface. Environ. Sci. Technol. 2010, 44, 170–176. [Google Scholar] [CrossRef]
- Missana, T.; Maffiotte, C.; García-Gutiérrez, M. Surface reactions kinetics between nanocrystalline magnetite and uranyl. J. Colloid Interface Sci. 2003, 261, 154–160. [Google Scholar] [CrossRef]
- Yuan, K.; Ilton, E.S.; Antonio, M.R.; Li, Z.; Cook, P.J.; Becker, U. Electrochemical and Spectroscopic Evidence on the One-Electron Reduction of U(VI) to U(V) on Magnetite. Environ. Sci. Technol. 2015, 49, 6206–6213. [Google Scholar] [CrossRef]
- Brookshaw, D.R.; Pattrick, R.A.D.; Bots, P.; Law, G.T.W.; Lloyd, J.R.; Mosselmans, J.F.W.; Vaughan, D.J.; Dardenne, K.; Morris, K. Redox interactions of Tc(VII), U(VI) and Np(V) with microbially reduced biotite and chlorite. Environ. Sci. Technol. 2015, 79, 13139–13148. [Google Scholar] [CrossRef]
- Begg, J.D.; Zavarin, M.; Zhao, P.; Tumey, S.J.; Powell, B.; Kersting, A.B. Pu(V) and Pu(IV) sorption to montmorillonite. Environ. Sci. Technol. 2013, 47, 5146–5153. [Google Scholar] [CrossRef]
- Zhao, P.; Begg, J.D.; Zavarin, M.; Tumey, S.J.; Williams, R.; Dai, Z.R.; Kips, R.; Kersting, A.B. Plutonium(IV) and (V) Sorption to Goethite at Sub-Femtomolar to Micromolar Concentrations: Redox Transformations and Surface Precipitation. Environ. Sci. Technol. 2016, 50, 6948–6956. [Google Scholar] [CrossRef]
- Law, G.T.W.; Geissler, A.; Lloyd, J.R.; Livens, F.R.; Boothman, C.; Begg, J.D.C.; Denecke, M.A.; Rothe, J.; Dardenne, K.; Burke, I.T.; et al. Geomicrobiological Redox Cycling of the Transuranic Element Neptunium. Environ. Sci. Technol. 2010, 44, 8924–8929. [Google Scholar] [CrossRef]
- Singer, D.M.; Chatman, S.M.; Ilton, E.S.; Rosso, K.M.; Banfield, J.F.; Waychunas, G.A. U(VI) Sorption and Reduction Kinetics on the Magnetite (111) Surface. Environ. Sci. Technol. 2012, 46, 3821–3830. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2)—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; U.S. Geological Survey: Denver, CO, USA, 1999; p. 312.
- Van Veelen, A.; Bargar, J.R.; Law, G.T.W.; Brown, G.E.; Wogelius, R.A. Uranium Immobilization and Nanofilm Formation on Magnesium-Rich Minerals. Environ. Sci. Technol. 2016, 50, 3435–3443. [Google Scholar] [CrossRef]
- Mosselmans, J.F.W.; Quinn, P.D.; Dent, A.J.; Cavill, S.A.; Moreno, S.D.; Peach, A.; Leicester, P.J.; Keylock, S.J.; Gregory, S.R.; Atkinson, K.D.; et al. I18—The microfocus spectroscopy beamline at the Diamond Light Source. J. Synchrotron Radiat. 2009, 16, 818–824. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Ankudinov, A.L.; Ravel, B.; Rehr, J.J.; Conradson, S.D. Real-space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Phys. Rev. B 1998, 58, 7565–7576. [Google Scholar] [CrossRef]
- Finch, R.J.; Cooper, M.A.; Hawthorne, F.C.; Ewing, R.C. The crystal structure of schoepite, [(UO2)8 O2(OH)12](H2O)12. Can. Mineral. 1996, 34, 1071–1088. [Google Scholar]
- Fleet, M. The structure of magnetite. Acta Crystallogr. Sect. B 1981, 37, 917–920. [Google Scholar] [CrossRef]
- Zachariasen, W.H. Crystal chemical studies of the 5f-Series of elements. XII. New compounds representing known structure types. Acta Crystallogr. 1949, 2, 388–390. [Google Scholar] [CrossRef]
- Downward, L.; Booth, C.H.; Lukens, W.W.; Bridges, F. A variation of the F-test for determining statistical relevance of particular parameters in EXAFS fits. AIP Conf. Proc. 2006, 882, 129–131. [Google Scholar] [CrossRef]
- Kvashnina, K.O.; Kvashnin, Y.O.; Butorin, S.M. Role of resonant inelastic X-ray scattering in high-resolution core-level spectroscopy of actinide materials. J. Electron Spectrosc. Relat. Phenom. 2014, 194, 27–36. [Google Scholar] [CrossRef]
- Denecke, M.A.; Dardenne, K.; Marquardt, C.M. Np(IV)/Np(V) valence determinations from Np L3 edge XANES/EXAFS. Talanta 2005, 65, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Ilton, E.S.; Bagus, P.S. XPS determination of uranium oxidation states. Surf. Interface Anal. 2011, 43, 1549–1560. [Google Scholar] [CrossRef]
- Ilton, E.S.; Boily, J.F.; Bagus, P.S. Beam induced reduction of U(VI) during X-ray photoelectron spectroscopy: The utility of the U4f satellite structure for identifying uranium oxidation states in mixed valence uranium oxides. Surf. Sci. 2007, 601, 908–916. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Veal, B.W.; Lam, D.J.; Diamond, H.; Hoekstra, H.R. X-ray photoelectron-spectroscopy study of oxides of the transuranium elements Np, Pu, Am, Cm, Bk, and Cf. Phys. Rev. B 1977, 15, 2929–2942. [Google Scholar] [CrossRef]
- Allen, G.C.; Crofts, J.A.; Curtis, M.T.; Tucker, P.M.; Chadwick, D.; Hampson, P.J. X-Ray photoelectron spectroscopy of some uranium oxide phases. J. Chem. Soc. Dalton Trans. 1974, 1296–1301. [Google Scholar] [CrossRef]
- Seibert, A.; Gouder, T.; Huber, F. Reaction of neptunium with molecular and atomic oxygen: Formation and stability of surface oxides. J. Nucl. Mater. 2009, 389, 470–478. [Google Scholar] [CrossRef]
- Seibert, A.; Gouder, T.; Huber, F. Interaction of PuO2 thin films with water. Radiochim. Acta 2010, 98, 647–654. [Google Scholar] [CrossRef]
- Belli, K.M.; DiChristina, T.J.; Van Cappellen, P.; Taillefert, M. Effects of aqueous uranyl speciation on the kinetics of microbial uranium reduction. Geochim. Cosmochim. Acta 2015, 157, 109–124. [Google Scholar] [CrossRef]
- Regenspurg, S.; Schild, D.; Schäfer, T.; Huber, F.; Malmström, M.E. Removal of uranium(VI) from the aqueous phase by iron(II) minerals in presence of bicarbonate. Appl. Geochem. 2009, 24, 1617–1625. [Google Scholar] [CrossRef]
- Missana, T.; García-Gutiérrez, M.; Fernńdez, V. Uranium (VI) sorption on colloidal magnetite under anoxic environment: Experimental study and surface complexation modelling. Geochim. Cosmochim. Acta 2003, 67, 2543–2550. [Google Scholar] [CrossRef]
- Petitto, S.C.; Tanwar, K.S.; Ghose, S.K.; Eng, P.J.; Trainor, T.P. Surface structure of magnetite (111) under hydrated conditions by crystal truncation rod diffraction. Surf. Sci. 2010, 604, 1082–1093. [Google Scholar] [CrossRef]
- Chadwick, D. Uranium 4f binding energies studied by X-ray photoelectron spectroscopy. Chem. Phys. Lett. 1973, 21, 291–294. [Google Scholar] [CrossRef]
- Wersin, P.; Hochella Jr, M.F.; Persson, P.; Redden, G.; Leckie, J.O.; Harris, D.W. Interaction between aqueous uranium (VI) and sulfide minerals: Spectroscopic evidence for sorption and reduction. Geochim. Cosmochim. Acta 1994, 58, 2829–2843. [Google Scholar] [CrossRef]
- Hennig, C.; Ikeda-Ohno, A.; Tsushima, S.; Scheinost, A.C. The sulfate coordination of Np(IV), Np(V), and Np(VI) in aqueous solution. Inorg. Chem. 2009, 48, 5350–5360. [Google Scholar] [CrossRef] [PubMed]
- Husar, R.; Hübner, R.; Hennig, C.; Martin, P.M.; Chollet, M.; Weiss, S.; Stumpf, T.; Zänker, H.; Ikeda-Ohno, A. Intrinsic formation of nanocrystalline neptunium dioxide under neutral aqueous conditions relevant to deep geological repositories. Chem. Commun. 2015, 51, 1301–1304. [Google Scholar] [CrossRef]
- Cakir, P.; Eloirdi, R.; Huber, F.; Konings, R.J.M.; Gouder, T. Surface reduction of neptunium dioxide and uranium mixed oxides with plutonium and thorium by photocatalytic reaction with ice. J. Phys. Chem. C 2015, 119, 1330–1337. [Google Scholar] [CrossRef]
- Teterin, A.Y.; Maslakov, K.I.; Teterin, Y.A.; Kalmykov, S.N.; Ivanov, K.E.; Vukcevic, L.; Khasanova, A.B.; Shcherbina, N.S. Interaction of neptunyl with goethite (alpha-FeOOH), maghemite (gamma-Fe2O3), and hematite (alpha-Fe2O3) in water as probed by X-ray photoelectron spectroscopy. Russ. J. Inorg. Chem. 2006, 51, 1937–1944. [Google Scholar] [CrossRef]
- Cox, L.E.; Farr, J.D. 4f binding-energy shifts of the light-actinide dioxides and tetrafluorides. Phys. Rev. B 1989, 39, 11142–11145. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, Y.; Tian, G. Np(v) complexation with carbonate in aqueous solutions studied by spectrophotometric titration at various temperatures. Dalton Trans. 2016, 45, 2681–2685. [Google Scholar] [CrossRef]
- Dong, W.; Brooks, S.C. Determination of the Formation Constants of Ternary Complexes of Uranyl and Carbonate with Alkaline Earth Metals (Mg2+, Ca2+, Sr2+, and Ba2+) Using Anion Exchange Method. Environ. Sci. Technol. 2006, 40, 4689–4695. [Google Scholar] [CrossRef]
- Wander, M.C.F.; Kerisit, S.; Rosso, K.M.; Schoonen, M.A.A. Kinetics of triscarbonato uranyl reduction by aqueous ferrous iron: A theoretical study. J. Phys. Chem. A 2006, 110, 9691–9701. [Google Scholar] [CrossRef]
- García Flores, H.G.; Roussel, P.; Moore, D.P.; Pugmire, D.L. Characterization and stability of thin oxide films on plutonium surfaces. Surf. Sci. 2011, 605, 314–320. [Google Scholar] [CrossRef]
- Marquardt, C.M.; Seibert, A.; Artinger, R.; Denecke, M.A.; Kuczewski, B.; Schild, D.; Fanghänel, T. The redox behaviour of plutonium in humic rich groundwater. Radiochim. Acta 2004, 92, 617–623. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Sample | Scattering Path | CN | R (Å) | σ2 (Å2) | S02 | R-factor |
|---|---|---|---|---|---|---|
| U MOPS 1 | U-Oax | 2 2 | 1.788 (4) | 0.0045 (5) | 1 2 | 0.007 |
| Edge step: 1.7 | U-Oeq | 3 2 | 2.205 (7) | 0.0051 (9) | ||
| U-Oeq | 3 2 | 2.408 (8) | 0.0035 (9) | |||
| U-Fe | 0.5 2 | 3.49 (3) | 0.006 (3) | |||
| U-Oax (MS) | 6 2 | 3.575 2 | 0.0089 2 | |||
| U NaHCO3 | U-Oax | 2 2 | 1.80 (7) | 0.010 (8) | 1 2 | 0.01 |
| Edge step: 0.039 | U-Oeq | 5 2 | 2.35 (13) | 0.014 (11) | ||
| Np MOPS | Np-O | 8 2 | 2.34 (4) | 0.009 (2) | 12 | 0.03 |
| Edge step: 0.013 | ||||||
| Np NaHCO3 | Np-O | 8 2 | 2.32 (3) | 0.011 (2) | 1 2 | 0.03 |
| Edge step: 0.018 |
| Fe(II) | Fe(III) | Pre-peak | Satellite (Fe(III)) | U(IV) | U(VI) | Pu(III) | Pu(IV) | Np(IV) | |
|---|---|---|---|---|---|---|---|---|---|
| U MOPS-1 | 26.5 | 67.3 | 0.7 | 5.5 | 43.8 | 56.2 | - | - | - |
| U MOPS-2 | 23.6 | 71.2 | 1.0 | 4.2 | 15.5 | 84.5 | - | - | - |
| U NaHCO3 | 29.4 | 65.5 | 1.0 | 4.1 | 37.5 | 62.5 | - | - | - |
| Np/Pu MOPS | 27.5 | 66.5 | 1.0 | 5.0 | - | - | 65.6 | 34.4 | 100 |
| Np/Pu NaHCO3 | 26.9 | 67.6 | 0.9 | 4.6 | - | - | 53.0 | 47.0 | 100 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bots, P.; van Veelen, A.; Mosselmans, J.F.W.; Muryn, C.; Wogelius, R.A.; Morris, K. Neptunium(V) and Uranium(VI) Reactions at the Magnetite (111) Surface. Geosciences 2019, 9, 81. https://doi.org/10.3390/geosciences9020081
Bots P, van Veelen A, Mosselmans JFW, Muryn C, Wogelius RA, Morris K. Neptunium(V) and Uranium(VI) Reactions at the Magnetite (111) Surface. Geosciences. 2019; 9(2):81. https://doi.org/10.3390/geosciences9020081
Chicago/Turabian StyleBots, Pieter, Arjen van Veelen, J. Frederick W. Mosselmans, Christopher Muryn, Roy A. Wogelius, and Katherine Morris. 2019. "Neptunium(V) and Uranium(VI) Reactions at the Magnetite (111) Surface" Geosciences 9, no. 2: 81. https://doi.org/10.3390/geosciences9020081
APA StyleBots, P., van Veelen, A., Mosselmans, J. F. W., Muryn, C., Wogelius, R. A., & Morris, K. (2019). Neptunium(V) and Uranium(VI) Reactions at the Magnetite (111) Surface. Geosciences, 9(2), 81. https://doi.org/10.3390/geosciences9020081