Volcanic Plume Impact on the Atmosphere and Climate: O- and S-Isotope Insight into Sulfate Aerosol Formation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
| Ch1: | |
| Ch2: | |
| Ch3: | |
| Ch4: |
2. Methods
2.1. Volcanic SO2 and H2S Sampling
2.2. Sulfate Aerosol Sampling
2.3. Sulfate Chemical Composition and Preparation
- -
- BaSO4: For about 30 years (e.g., [36,45]) sulfates were transformed into highly insoluble BaSO4 by adding BaCl2 in the leachate obtained from different kind of samples such as ash or filters. However, when precipitated from a multi-anion solution, barite (BaSO4) can occlude impurities such as nitrate, which can introduce an analytical bias in the measurement of O-isotopes [46]. For this reason, it is of paramount importance to purify the collected sulfate and make sure that no nitrate remains in the leachate. In order to purify barite, Bao et al. [47] developed the DDARP (diethylenetriaminepentaacetic acid dissolution and re-precipitation) method, while more recently, Le Gendre et al. [48] worked out a Resin Method for Sulfate Extraction and Purification (RMSEP), that passes the leachate solution through an anionic exchange resin, allowing the purification and concentration of sulfate. Then, by adding BaCl2, pure BaSO4 is precipitated.
- -
- Ag2SO4: Via anionic exchange resins, sulfate from the leachate is converted into Na2SO4. Then Ag2SO4 is produced by passing the Na2SO4 through another resin conditioned into Ag+ [49].
- -
2.4. Oxygen Isotope Measurements
2.5. Sulfur Isotope Measurements
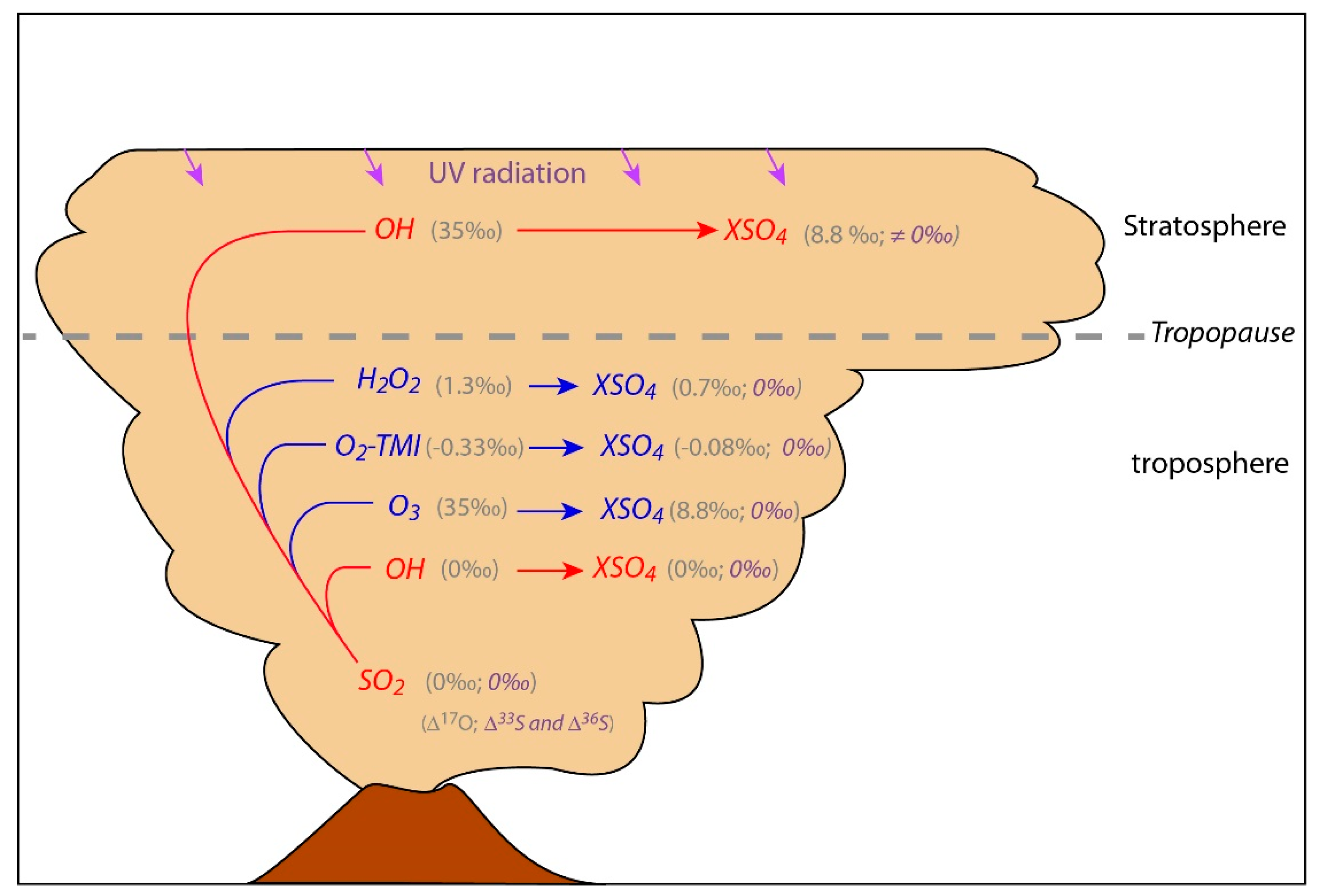
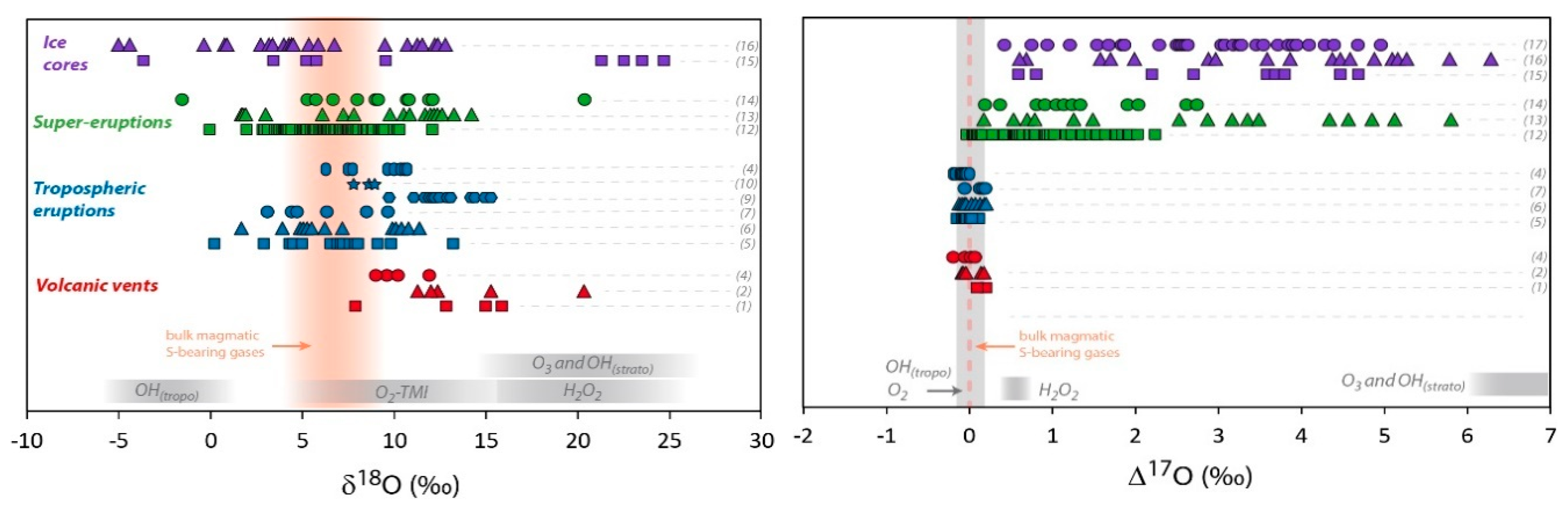
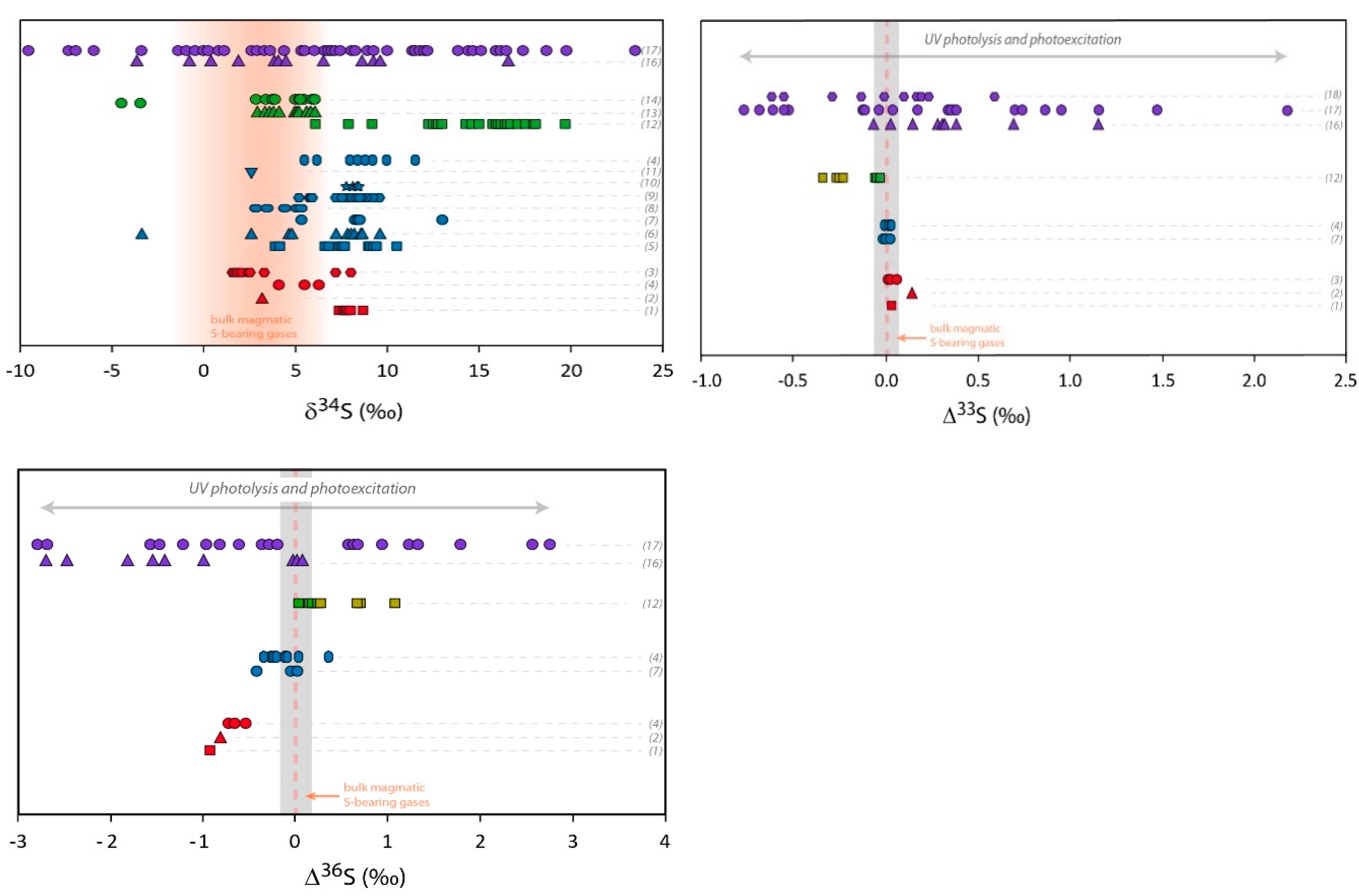
3. Volcanic Sulfate Formation
3.1. Isotopic Composition of S-Bearing Gases
3.2. High Temperature Primary Sulfates
3.3. Tropospheric Secondary Sulfates
3.4. Stratospheric Secondary Sulfates
3.5. Ash Layer from Large Caldera-Forming Eruptions (Super-Eruptions)
4. Summary and Conclusions
- -
- High temperature chemistry (including sulfate aerosol formation) is assessed by equilibrium thermodynamic models, but at volcanic vents the plume cools down and dilutes very quickly, therefore modeling using a kinetic approach should be more appropriate.
- -
- The isotopic approach alone can hardly differentiate between different possible mass-dependent processes responsible for sulfate formation. In volcanic plumes, sulfates can be generated by oxidation channels such as OH or O2-TMI oxidation, but the exploration of other possible oxidation processes is required. The role played by halogens and OH radicals generated from ash particles in the SO2 oxidation needs to be quantified as in volcanic plumes they may play a more preponderant role than expected. This would improve our understanding of sulfate aerosol formation in a relatively particle-dense plume or cloud.
- -
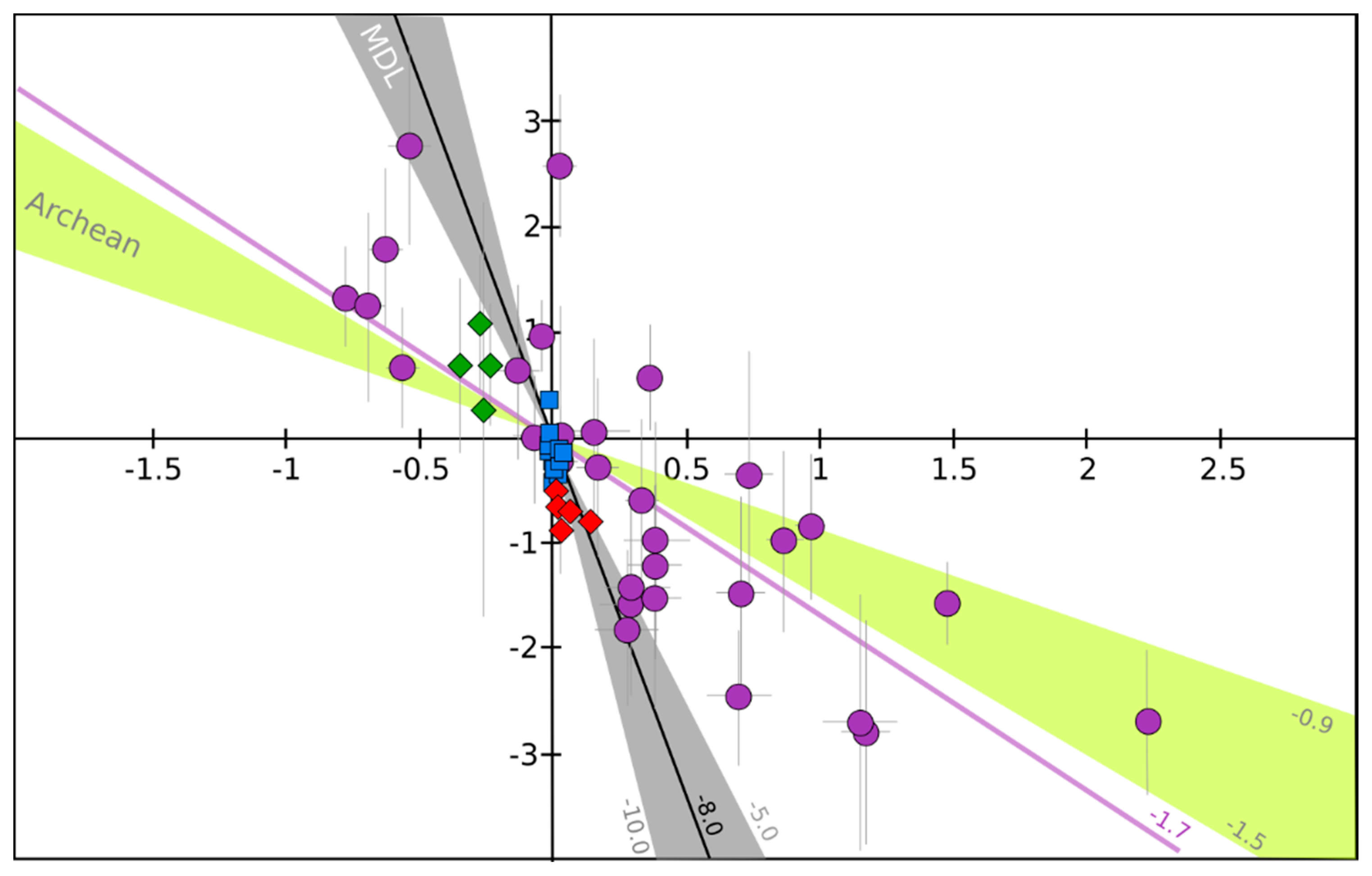
- In the stratosphere, the low Δ17O sulfates are not totally understood yet. Oxidation channels other than oxidation via OH need to be explored. Furthermore, the fact that in ice-cores the evolution of Δ33S (from negative to positive) and Δ36S (from positive to negative) is recorded during a single volcanic event on a year timescale, while the SO2 oxidation in the stratosphere takes a few months after an eruption, is still unexplained.
- -
- The impact of super-eruptions on the atmosphere and more specifically on the tropopause needs to be explored in more detail in order to better constrain the potential chemical fluxes between the troposphere and the stratosphere during such an event. This would have an impact on the sulfate aerosol formation and on the atmospheric and climatic impact of super-eruptions in general.
Acknowledgments
Conflicts of Interest
References
- Andres, R.J.; Kasgnoc, A.D. A time-averaged inventory of subaerial volcanic sulfur emissions. J. Geophys. Res. Atmos. 1998, 103, 25251–25261. [Google Scholar] [CrossRef]
- Graf, H.-F.; Feichter, J.; Langmann, B. Volcanic sulfur emissions: Estimates of source strength and its contribution to the global sulfate distribution. J. Geophys. Res. Atmos. 1997, 102, 10727–10738. [Google Scholar] [CrossRef]
- Textor, C.; Graf, H.-F.; Timmreck, C.; Robock, A. Emissions from volcanoes. In Emissions of Atmospheric Trace Compounds; Advances in Global Change Research; Springer: Dordrecht, The Netherlands, 2004; pp. 269–303. ISBN 978-90-481-6605-3. [Google Scholar]
- Delmelle, P.; Stix, J. Volcanic gases. In Encyclopedia of Volcanoes; Sigurdsson, H., Houghton, B., McNutt, S.R., Rymer, H., Stix, J., Eds.; Elsevier: New York, NY, USA, 2000; pp. 803–815. [Google Scholar]
- Oppenheimer, C.; Scaillet, B.; Martin, R.S. Sulfur Degassing From Volcanoes: Source Conditions, Surveillance, Plume Chemistry and Earth System Impacts. Rev. Mineral. Geochem. 2011, 73, 363–421. [Google Scholar] [CrossRef]
- Haywood, J.; Boucher, O. Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: A review. Rev. Geophys. 2000, 38, 513–543. [Google Scholar] [CrossRef]
- Robock, A. Volcanic eruption and climate. Rev. Geophys. 2000, 38, 191–219. [Google Scholar] [CrossRef]
- Hansen, J.E.; Wang, W.-C.; Lacis, A.A. Mount Agung Eruption Provides Test of a Global Climatic Perturbation. Science 1978, 199, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.E.; Wilson, H.; Jones, P.D.; Christy, J.R.; Folland, C.K. The impact of mount pinatubo on world-wide temperatures. Int. J. Climatol. 1996, 16, 487–497. [Google Scholar] [CrossRef]
- Parker, D.E.; Brownscombe, J.L. Stratospheric warming following the El Chichón volcanic eruption. Nature 1983, 301, 406–408. [Google Scholar] [CrossRef]
- Rampino, M.R.; Self, S. Historic Eruptions of Tambora (1815), Krakatau (1883), and Agung (1963), their Stratospheric Aerosols, and Climatic Impact. Quat. Res. 1982, 18, 127–143. [Google Scholar] [CrossRef]
- Farquhar, J.; Savarino, J.; Airieau, S.; Thiemens, M.H. Observation of wavelength-sensitive mass-independent sulfur isotope effects during SO2 photolysis: Implications for the early atmosphere. Geophys. Res. 2001, 106, 32829–32839. [Google Scholar] [CrossRef]
- Farquhar, J.; Bao, H.; Thiemens, M. Atmospheric Influence of Earth’s Earliest Sulfur Cycle. Science 2000, 289, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Thiemens, M.H. History and applications of mass-independent isotope effects. Annu. Rev. Earth Planet. Sci. 2006, 34, 217–262. [Google Scholar] [CrossRef]
- Thiemens, M.H.; Heidenreich, J.E. The Mass-Independent Fractionation of Oxygen: A Novel Isotope Effect and Its Possible Cosmochemical Implications. Science 1983, 219, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, J.; Wing, B.A. Multiple sulfur isotopes and the evolution of the atmosphere. Earth Planet. Sci. Lett. 2003, 213, 1–13. [Google Scholar] [CrossRef]
- Rumble, D.; Miller, M.F.; Franchi, I.A.; Greenwood, R.C. Oxygen three-isotope fractionation lines in terrestrial silicate minerals: An inter-laboratory comparison of hydrothermal quartz and eclogitic garnet. Geochim. Cosmochim. Acta 2007, 71, 3592–3600. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Savarino, J.; Lee, C.C.W.; Thiemens, M.H. Laboratory oxygen isotopic study of sulfur (IV) oxidation: Origin of the mass-independent oxygen isotopic anomaly in atmospheric sulfates and sulfate mineral deposits on Earth. J. Geophys. Res. Atmos. 2000, 105, 29079–29088. [Google Scholar] [CrossRef]
- Martin, E.; Bekki, S.; Ninin, C.; Bindeman, I. Volcanic sulfate aerosol formation in the troposphere. J. Geophys. Res. Atmos. 2014, 119, 12:660–12:673. [Google Scholar] [CrossRef]
- Ono, S.; Whitehill, A.R.; Lyons, J.R. Contribution of isotopologue self-shielding to sulfur massindependent fractionation during sulfur dioxide photolysis. Ournal Geophys. Res. Atmos. 2013, 118, 2444–2454. [Google Scholar] [CrossRef]
- Whitehill, A.R.; Xie, C.; Hu, X.; Xie, D.; Guo, H.; Ono, S. Vibronic origin of sulfur mass-independent isotope effect in photoexcitation of SO2 and the implications to the early earth’s atmosphere. Proc. Natl. Acad. Sci. USA 2013, 110, 17697–17702. [Google Scholar] [CrossRef] [PubMed]
- Giggenbach, W.F. A simple method for the collection and analysis of volcanic gas samples. Bull. Volcanol. 1975, 39, 132–145. [Google Scholar] [CrossRef]
- Giggenbach, W.F.; Goguel, R.L. Methods for the Collection and Analysis of Geothermal and Volcanic Water and Gas Samples; Chemistry Division, Department of Scientific and Industrial Research: Petone, New Zealand, 1989; p. 53.
- De Moor, J.M.; Fischer, T.P.; Sharp, Z.D.; King, P.L.; Wilke, M.; Botcharnikov, R.E.; Cottrell, E.; Zelenski, M.; Marty, B.; Klimm, K.; et al. Sulfur degassing at Erta Ale (Ethiopia) and Masaya (Nicaragua) volcanoes: Implications for degassing processes and oxygen fugacities of basaltic systems: Sulfur Degassing at Basaltic Volcanoes. Geochem. Geophys. Geosyst. 2013, 14, 4076–4108. [Google Scholar] [CrossRef]
- Menyailov, I.A.; Nikitina, L.P.; Shapar, V.N.; Pilipenko, V.P. Temperature increase and chemical change of fumarolic gases at Momotombo Volcano, Nicaragua, in 1982–1985: Are these indicators of a possible eruption? J. Geophys. Res. Solid Earth 1986, 91, 12199–12214. [Google Scholar] [CrossRef]
- Goff, F.; Janik, C.J.; Delgado, H.; Werner, C.; Counce, D.; Stimac, J.A.; Siebe, C.; Love, S.P.; Williams, S.N.; Fischer, T.; et al. Geochemical surveillance of magmatic volatiles at Popocatépetl volcano, Mexico. GSA Bull. 1998, 110, 695–710. [Google Scholar] [CrossRef]
- Ohba, T.; Nogami, K.; Hirabayashi, J.-I.; Mori, T. Isotopic fractionation of SO2 and H2S gases during the absorption by KOH solution, with the application to volcanic gas monitoring at Miyakejima Island, Japan. Geochem. J. 2008, 42, 119–131. [Google Scholar] [CrossRef]
- Mather, T.A.; Pyle, D.M.; Heaton, T.H.E. Investigation of the use of filter packs to measure the sulphur isotopic composition of volcanic sulphur dioxide and the sulphur and oxygen isotopic composition of volcanic sulphate aerosol. Atmos. Environ. 2008, 42, 4611–4618. [Google Scholar] [CrossRef]
- Le Gendre, E. Étude des Anomalies Isotopiques de L’oxygène Etdusoufre Dans les Sulfates D’origine Volcanique et Anthropique; UPMC: Pittsburgh, PA, USA, 2016. [Google Scholar]
- Mather, T.A.; McCabe, J.R.; Rai, V.K.; Thiemens, M.H.; Pyle, D.M.; Heaton, T.H.E.; Sloane, H.J.; Fern, G.R. Oxygen and sulfur isotopic composition of volcanic sulfate aerosol at the point of emission. J. Geophys. Res. Atmos. 2006, 111, D18205. [Google Scholar] [CrossRef]
- Armienta, M.A.; De la Cruz-Reyna, S.; Soler, A.; Cruz, O.; Ceniceros, N.; Aguayo, A. Chemistry of ash-leachates to monitor volcanic activity: An application to Popocatépetl volcano, central Mexico. Appl. Geochem. 2010, 25, 1198–1205. [Google Scholar] [CrossRef]
- Martin, E.; Bindeman, I. Mass-independent isotopic signatures of volcanic sulfate from three supereruption ash deposits in Lake Tecopa, California. Earth Planet. Sci. Lett. 2009, 282, 102–114. [Google Scholar] [CrossRef]
- Risacher, F.; Alonso, H. Geochemistry of ash leachates from the 1993 Lascar eruption, northern Chile. Implication for recycling of ancient evaporites. J. Volcanol. Geotherm. Res. 2001, 109, 319–337. [Google Scholar] [CrossRef]
- Bao, H.; Thiemens, M.H.; Loope, D.B.; Yuan, X.-L. Sulfate oxygen-17 anomaly in an Oligocene ash bed in mid-North America: Was it the dry fogs? Geophys. Res. Lett. 2003, 30, 11–14. [Google Scholar] [CrossRef]
- Van Stempvoort, D.R.; Krouse, H.R. Controls of d18O in sulfate: Review of experimental data and application to specific environments. In Environmental Geochemistry of Sulfide Oxidation; Alpers, C.N., Blowes, D.W., Eds.; American Chemical Society: Washington, DC, USA, 1994; Volume 550. [Google Scholar]
- Witham, C.S.; Oppenheimer, C.; Horwell, C.J. Volcanic ash-leachates: A review and recommendations for sampling methods. J. Volcanol. Geotherm. Res. 2005, 141, 299–326. [Google Scholar] [CrossRef]
- Botter, C. Utomated Single-Particle SEM/EDS Analysis of Volcanic Aerosols from Stromboli, Aeolian Arc, Italy; Mineralogical Characterization of the PM10 Fraction; University of Fribourg: Fribourg, Switzerland, 2011. [Google Scholar]
- Botter, C.; Meier, M.; Wiedenmann, D.; Grobety, B.; Ricci, T. Single Particle Analysis of Volcanic Aerosols from Stromboli, Italy. In Proceedings of the Cities on Volcanoes 6 (COV6), Tenerife, Spain, 31 May–4 June 2010. [Google Scholar]
- Toutain, J.; Quisefit, J.; Briole, P.; Aloupogiannis, P.; Blanc, P.; Robaye, G. Mineralogy and chemistry of solid aerosols emitted from Mount Etna. Geochem. J. 1995, 29, 163–173. [Google Scholar] [CrossRef]
- Stoiber, R.E.; Rose, W.I. The Geochemistry of Central American Volcanic Gas Condensates. GSA Bull. 1970, 81, 2891–2912. [Google Scholar] [CrossRef]
- Symonds, R.B.; Reed, M.H.; Rose, W.I. Origin, speciation, and fluxes of trace-element gases at Augustine volcano, Alaska: Insights into magma degassing and fumarolic processes. Geochim. Cosmochim. Acta 1992, 56, 633–657. [Google Scholar] [CrossRef]
- Quisefit, J.P.; Bergametti, G.; Tedesco, D.; Pinart, J.; Colin, J.L. Origin of particulate potassium in Mt Etna emissions before and during the 1983 eruption. J. Volcanol. Geotherm. Res. 1988, 35, 111–119. [Google Scholar] [CrossRef]
- Ayris, P.M.; Lee, A.F.; Wilson, K.; Kueppers, U.; Dingwell, D.B.; Delmelle, P. SO2 sequestration in large volcanic eruptions: High-temperature scavenging by tephra. Geochim. Cosmochim. Acta 2013, 110, 58–69. [Google Scholar] [CrossRef]
- Van Stempvoort, D.R.; Reardon, E.J.; Fritz, P. Fractionation of sulfur and oxygen isotopes in sulfate by soil sorption. Geochim. Cosmochim. Acta 1990, 54, 2817–2826. [Google Scholar] [CrossRef]
- Michalski, G.; Kasem, M.; Rech, J.A.; Adieu, S.; Showers, W.S.; Genna, B.; Thiemens, M. Uncertainties in the oxygen isotopic composition of barium sulfate induced by coprecipitation of nitrate. Rapid Commun. Mass Spectrom. 2008, 22, 2971–2976. [Google Scholar] [CrossRef] [PubMed]
- Bao, H. Purifying barite for oxygen isotope measurement by dissolution and reprecipitation in a chelating solution. Anal. Chem. 2006, 78, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Le Gendre, E.; Martin, E.; Villemant, B.; Cartigny, P.; Assayag, N. A simple and reliable anion-exchange resin method for sulfate extraction and purification suitable for multiple O- and S-isotope measurements: Anion-exchange method for multiple O- and S-isotope analysis. Rapid Commun. Mass Spectrom. 2017, 31, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Schauer, A.J.; Kunasek, S.A.; Sofen, E.D.; Erbland, J.; Savarino, J.; Johnson, B.W.; Amos, H.M.; Shaheen, R.; Abaunza, M.; Jackson, T.L.; et al. Oxygen isotope exchange with quartz during pyrolysis of silver sulfate and silver nitrate: Oxygen isotope exchange during pyrolysis of Ag2SO4 and AgNO3. Rapid Commun. Mass Spectrom. 2012, 26, 2151–2157. [Google Scholar] [CrossRef] [PubMed]
- Pepkowitz, L.; Shirley, E. microdetection of sulfur. Anal. Chem. 1951, 23, 1709–1710. [Google Scholar] [CrossRef]
- Farquhar, J.; Peters, M.; Johnston, D.T.; Strauss, H.; Masterson, A.; Wiechert, U.; Kaufman, A.J. Isotopic evidence for Mesoarchaean anoxia and changing atmospheric sulphur chemistry. Nature 2007, 449, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Rafter, T.A. Oxygen isotopic composition of sulfates. Part I. A method for the extraction of oxygen and its quantitative conversion to carbon dioxide for isotope radiation measurements. N. Z. J. Sci. 1967, 10, 493–510. [Google Scholar]
- Mizutani, Y. An improvement in the carbon-reduction method for the oxygen isotopic analysis of sulphates. Geochem. J. 1971, 5, 69–77. [Google Scholar] [CrossRef]
- Bao, H.; Thiemens, M.H. Generation of O2 from BaSO4 using a CO2-laser fluorination system for simultaneous analysis of d18O and d17O. Anal. Chem. 2000, 72, 4029–4032. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Schauer, A.J.; Kunasek, S.A.; Sofen, E.D.; Erbland, J.; Savarino, J.; Allman, D.J.; Sletten, R.S.; Alexander, B. Analysis of oxygen-17 excess of nitrate and sulfate at sub-micromole levels using the pyrolysis method: Analysis of oxygen-17 excess of nitrate and sulfate. Rapid Commun. Mass Spectrom. 2013, 27, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Savarino, J.; Alexander, B.; Darmohusodo, V.; Thiemens, M.H. Sulfur and Oxygen Isotope Analysis of Sulfate at Micromole Levels Using a Pyrolysis Technique in a Continuous Flow System. Anal. Chem. 2001, 73, 4457–4462. [Google Scholar] [CrossRef] [PubMed]
- Au Yang, D.; Landais, G.; Assayag, N.; Widory, D.; Cartigny, P. Improved analysis of micro- and nanomole-scale sulfur multi-isotope compositions by gas source isotope ratio mass spectrometry: Improved analysis of S isotopes at micro/nanomole levels by IRMS. Rapid Commun. Mass Spectrom. 2016, 30, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.S.; Sawyer, G.M.; Spampinato, L.; Salerno, G.G.; Ramirez, C.; Ilyinskaya, E.; Witt, M.L.I.; Mather, T.A.; Watson, I.M.; Phillips, J.C.; et al. A total volatile inventory for Masaya Volcano, Nicaragua. J. Geophys. Res. 2010, 115. [Google Scholar] [CrossRef]
- Allen, A.G.; Oppenheimer, C.; Ferm, M.; Baxter, P.J.; Horrocks, L.A.; Galle, B.; McGonigle, A.J.S.; Duffell, H.J. Primary sulfate aerosol and associated emissions from Masaya Volcano, Nicaragua. J. Geophys. Res. Atmos. 2002, 107, 4682. [Google Scholar] [CrossRef]
- Cwiertny, D.M.; Young, M.A.; Grassian, V.H. Chemistry and Photochemistry of Mineral Dust Aerosol. Annu. Rev. Phys. Chem. 2008, 59, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Casadevall, T.J.; Moore, J.G. Chemistry and isotope ratios of sulfur in basalts and volcanic gases at Kilauea volcano, Hawaii. Geochim. Cosmochim. Acta 1982, 46, 729–738. [Google Scholar] [CrossRef]
- Allard, P. 13C/12C and 34S/32S ratios in magmatic gases from ridge volcanism in Afar. Nature 1979, 282, 56–58. [Google Scholar] [CrossRef]
- Sakai, H.; Gunnlaugsson, E.; Tòmasson, J.; Rouse, J.E. Sulfur isotope systematics in icelandic geothermal systems and influence of seawater circulation at Reykjanes. Geochim. Cosmochim. Acta 1980, 44, 1223–1231. [Google Scholar] [CrossRef]
- Goff, F.; McMurtry, G.M. Tritium and stable isotopes of magmatic waters. J. Volcanol. Geotherm. Res. 2000, 97, 347–396. [Google Scholar] [CrossRef]
- Taran, Y.; Gavilanes, J.C.; Cortés, A. Chemical and isotopic composition of fumarolic gases and the SO2 flux from Volcán de Colima, México, between the 1994 and 1998 eruptions. J. Volcanol. Geotherm. Res. 2002, 117, 105–119. [Google Scholar] [CrossRef]
- Sakai, H.; Matsubaya, O. Stable isotopic studies of japanese geothermal systems. Geothermics 1977, 5, 97–124. [Google Scholar] [CrossRef]
- Giggenbach, W. The chemical and isotopic composition of gas discharges from New Zealand andesitic volcanoes. Bull. Volcanol. 1982, 45, 253–255. [Google Scholar] [CrossRef]
- Williams, S.N.; Sturchio, N.C.; Calvache, V.M.L.; Mendez, F.R.; Londoño, C.A.; García, P.N. Sulfur dioxide from Nevado del Ruiz volcano, Colombia: Total flux and isotopic constraints on its origin. J. Volcanol. Geotherm. Res. 1990, 42, 53–68. [Google Scholar] [CrossRef]
- Richet, P.; Bottinga, Y.; Javoy, M. A Review of Hydrogen, Carbon, Nitrogen, Oxygen, Sulphur, and Chlorine Stable Isotope Fractionation Among Gaseous Molecules. Annu. Rev. Earth Planet. Sci. 1977, 5, 65–110. [Google Scholar] [CrossRef]
- Ohmoto, H.; Rye, R.O. Isotope of sulfur and carbon. In Geochemestry of Hydrothermal Deposits; Barnes, H.L., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 1979; pp. 509–567. [Google Scholar]
- Labidi, J.; Cartigny, P.; Birck, J.L.; Assayag, N.; Bourrand, J.J. Determination of multiple sulfur isotopes in glasses: A reappraisal of the MORB δ34S. Chem. Geol. 2012, 334, 189–198. [Google Scholar] [CrossRef]
- De Hoog, J.C.M.; Taylor, B.E.; van Bergen, M.J. Sulfur isotope systematics of basaltic lavas from Indonesia: Implications for the sulfur cycle in subduction zones. Earth Planet. Sci. Lett. 2001, 189, 237–252. [Google Scholar] [CrossRef]
- Martin, E.; Bindeman, I.; Grove, T. The origin of high-Mg magmas in Mt Shasta and Medicine Lake volcanoes, Cascade Arc (California): Higher and lower than mantle oxygen isotope signatures attributed to current and past subduction. Contrib. Miner. Petrol. 2011, 162, 945–960. [Google Scholar] [CrossRef]
- Eiler, J. Oxygen isotope variations of basaltic lavas and upper mantle rocks. In Reviews in Mineralogy and Geochemistry; Walley, J.W., Cole, D.R., Eds.; The Mineralogical Society of America: Washington, DC, USA, 2001; Volume 43, pp. 319–364. [Google Scholar]
- Bindeman, I.N.; Eiler, J.M.; Wing, B.A.; Farquhar, J. Rare sulfur and triple oxygen isotope geochemistry of volcanogenic sulfate aerosols. Geochim. Cosmochim. Acta 2007, 71, 2326–2343. [Google Scholar] [CrossRef]
- De Moor, J.M.; Fischer, T.P.; Hilton, D.R.; Hauri, E.; Jaffe, L.A.; Camacho, J.T. Degassing at Anatahan volcano during the May 2003 eruption: Implications from petrology, ash leachates, and SO2 emissions. J. Volcanol. Geotherm. Res. 2005, 146, 117–138. [Google Scholar] [CrossRef]
- Rye, R.O.; Luhr, J.F.; Wasserman, M.D. Sulfur and oxygen isotopic systematics of the 1982 eruptions of El Chichón Volcano, Chiapas, Mexico. J. Volcanol. Geotherm. Res. 1984, 23, 109–123. [Google Scholar] [CrossRef]
- Savarino, J.; Bekki, S.; Cole-Dai, J.H.; Thiemens, M.H. Evidence from sulfate mass independent oxygen isotopic compositions of dramatic changes in atmospheric oxidation following massive volcanic eruptions. J. Geophys. Res. Atmos. 2003, 108. [Google Scholar] [CrossRef]
- Baroni, M. Etude des Anomalies Isotopiques du Soufre et de L’oxygène Dans le Sulfate D’origine Volcanique Enregistré dans les Archives Glaciaires; Univeristé Joseph Fourier: Grenoble, France, 2006. [Google Scholar]
- Baroni, M.; Thiemens, M.H.; Delmas, R.J.; Savarino, J. Mass-Independent Sulfur Isotopic Compositions in Stratospheric Volcanic Eruptions. Science 2007, 315, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Savarino, J.; Cole-Dai, J.; Rai, V.K.; Thiemens, M.H. Anomalous sulfur isotope compositions of volcanic sulfate over the last millennium in Antarctic ice cores. J. Geophys. Res. Atmos. 2008, 113, D20112. [Google Scholar] [CrossRef]
- Gautier, E. Empreinte Isotopique et Histoire du Volcanisme Stratosphérique des 2600 Dernières Années, Enregistrées à Dome C, Antarctique; Université Grenoble Alpes: Grenoble, France, 2015. [Google Scholar]
- Cole-Dai, J.; Ferris, D.; Lanciki, A.; Savarino, J.; Baroni, M.; Thiemens, M.H. Cold decade (AD 1810–1819) caused by Tambora (1815) and another (1809) stratospheric volcanic eruption. Geophys. Res. Lett. 2009, 36. [Google Scholar] [CrossRef]
- Miyoshi, T.; Sakai, H.; Chiba, H. Experimental study of sulfur isotope fractionation factors between sulfate and sulfide in high temperature melts. Geochem. J. 1984, 18, 75–84. [Google Scholar] [CrossRef]
- Hoefs, J. Stable Isotope Geochemistry; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Farquhar, J.; Johnston, D.T.; Wing, B.A. Implications of conservation of mass effects on mass-dependent isotope fractionations: Influence of network structure on sulfur isotope phase space of dissimilatory sulfate reduction. Geochim. Cosmochim. Acta 2007, 71, 5862–5875. [Google Scholar] [CrossRef]
- Ono, S.; Wing, B.; Johnston, D.; Farquhar, J.; Rumble, D. Mass-dependent fractionation of quadruple stable sulfur isotope system as a new tracer of sulfur biogeochemical cycles. Geochim. Cosmochim. Acta 2006, 70, 2238–2252. [Google Scholar] [CrossRef]
- Martin, R.S.; Mather, T.A.; Pyle, D.M. High-temperature mixtures of magmatic and atmospheric gases. Geochem. Geophys. Geosyst. 2006, 7, Q04006. [Google Scholar] [CrossRef]
- Roberts, T.J.; Braban, C.F.; Martin, R.S.; Oppenheimer, C.; Adams, J.W.; Cox, R.A.; Jones, R.L.; Griffiths, P.T. Modelling reactive halogen formation and ozone depletion in volcanic plumes. Chem. Geol. 2009, 263, 151–163. [Google Scholar] [CrossRef]
- Roberts, T.J.; Martin, R.S.; Jourdain, L. Reactive bromine chemistry in Mount Etna’s volcanic plume: The influence of total Br, high-temperature processing, aerosol loading and plume–air mixing. Atmos. Chem. Phys. 2014, 14, 11201–11219. [Google Scholar] [CrossRef]
- Gerlach, T.M. Volcanic sources of tropospheric ozone-depleting trace gases. Geochem. Geophys. Geosyst. 2004, 5. [Google Scholar] [CrossRef]
- Lee, C.C.-W.; Savarino, J.H.; Cachier, H.; Thiemens, M.H. Sulfur (32S, 33S, 34S, 36S) and oxygen (16O,17O,18O) isotopic ratios of primary sulfate produced from combustion processes. Tellus B 2002, 54, 193–200. [Google Scholar] [CrossRef]
- Belo, L.P.; Elliott, L.K.; Stanger, R.J.; Spörl, R.; Shah, K.V.; Maier, J.; Wall, T.F. High-Temperature Conversion of SO2 to SO3: Homogeneous Experiments and Catalytic Effect of Fly Ash from Air and Oxy-fuel Firing. Energy Fuels 2014, 28, 7243–7251. [Google Scholar] [CrossRef]
- Ono, S. Photochemistry of Sulfur Dioxide and the Origin of Mass-Independent Isotope Fractionation in Earth’s Atmosphere. Annu. Rev. Earth Planet. Sci. 2017, 45, 301–329. [Google Scholar] [CrossRef]
- Jenkins, K.A.; Bao, H. Multiple oxygen and sulfur isotope compositions of atmospheric sulfate in Baton Rouge, LA, USA. Atmos. Environ. 2006, 40, 4528–4537. [Google Scholar] [CrossRef]
- Lee, C.C.-W.; Thiemens, M.H. The δ17O and δ18O measurements of atmospheric sulfate from a coastal and high alpine region: A mass-independent isotopic anomaly. J. Geophys. Res. Atmos. 2001, 106, 17359–17373. [Google Scholar] [CrossRef]
- Li, X.; Bao, H.; Gan, Y.; Zhou, A.; Liu, Y. Multiple oxygen and sulfur isotope compositions of secondary atmospheric sulfate in a mega-city in central China. Atmos. Environ. 2013, 81, 591–599. [Google Scholar] [CrossRef]
- Carn, S.A.; Clarisse, L.; Prata, A.J. Multi-decadal satellite measurements of global volcanic degassing. J. Volcanol. Geotherm. Res. 2016, 311, 99–134. [Google Scholar] [CrossRef]
- Yang, K.; Krotkov, N.A.; Krueger, A.J.; Carn, S.A.; Bhartia, P.K.; Levelt, P.F. Improving retrieval of volcanic sulfur dioxide from backscattered UV satellite observations. Geophys. Res. Lett. 2009, 36, L03102. [Google Scholar] [CrossRef]
- Galeazzo, T.; Bekki, S.; Martin, E. Modeling the Pathways of Generation of O-MIF in Tropospheric Sulfates; GOLDSCHMIDT: Paris, France, 2017. [Google Scholar]
- Kroll, J.H.; Cross, E.S.; Hunter, J.F.; Pai, S.; Wallace, L.M.M.; Croteau, P.L.; Jayne, J.T.; Worsnop, D.R.; Heald, C.L.; Murphy, J.G.; et al. Atmospheric Evolution of Sulfur Emissions from Kı̅lauea: Real-Time Measurements of Oxidation, Dilution, and Neutralization within a Volcanic Plume. Environ. Sci. Technol. 2015, 49, 4129–4137. [Google Scholar] [CrossRef] [PubMed]
- Dupart, Y.; King, S.M.; Nekat, B.; Nowak, A.; Wiedensohler, A.; Herrmann, H.; David, G.; Thomas, B.; Miffre, A.; Rairoux, P.; et al. Mineral dust photochemistry induces nucleation events in the presence of SO2. Proc. Natl. Acad. Sci. USA 2012, 109, 20842–20847. [Google Scholar] [CrossRef] [PubMed]
- Vallyathan, V.; Shi, X.; Dalal, N.S.; Irr, W.; Castranova, V. Generation of Free Radicals from Freshly Fractured Silica Dust: Potential Role in Acute Silica-induced Lung Injury. Am. Rev. Respir. Dis. 1988, 138, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Konecny, R. Reactivity of Hydroxyl Radicals on Hydroxylated Quartz Surface. 1. Cluster Model Calculations. J. Phys. Chem. B 2001, 105, 6221–6226. [Google Scholar] [CrossRef]
- Narayanasamy, J.; Kubicki, J.D. Mechanism of Hydroxyl Radical Generation from a Silica Surface: Molecular Orbital Calculations. J. Phys. Chem. B 2005, 109, 21796–21807. [Google Scholar] [CrossRef] [PubMed]
- Von Glasow, R.; Sander, R.; Bott, A.; Crutzen, P.J. Modeling halogen chemistry in the marine boundary layer 2. Interactions with sulfur and the cloud-covered MBL. J. Geophys. Res. Atmos. 2002, 107, 4323. [Google Scholar] [CrossRef]
- Roberts, T.J.; Jourdain, L.; Griffiths, P.T.; Pirre, M. Re-evaluating the reactive uptake of HOBr in the troposphere with implications for the marine boundary layer and volcanic plumes. Atmos. Chem. Phys. 2014, 14, 11185–11199. [Google Scholar] [CrossRef]
- McCormick, M.P.; Thomason, L.W.; Trepte, C.R. Atmospheric effects of the Mt Pinatubo eruption. Nature 1995, 373, 399–404. [Google Scholar] [CrossRef]
- Castellano, E.; Becagli, S.; Jouzel, J.; Migliori, A.; Severi, M.; Steffensen, J.P.; Traversi, R.; Udisti, R. Volcanic eruption frequency over the last 45 ky as recorded in Epica-Dome C ice core (East Antarctica) and its relationship with climatic changes. Glob. Planet. Chang. 2004, 42, 195–205. [Google Scholar] [CrossRef]
- Cole-Dai, J.; Mosley-Thompson, E.; Wight, S.P.; Thompson, L.G. A 4100-year record of explosive volcanism from an East Antarctica ice core. J. Geophys. Res. Atmos. 2000, 105, 24431–24441. [Google Scholar] [CrossRef]
- Delmas, R.J.; Kirchner, S.; Palais, J.M.; Petit, J.-R. 1000 years of explosive volcanism recorded at the South Pole. Tellus B 1992, 44, 335–350. [Google Scholar] [CrossRef]
- Langway, C.C.; Osada, K.; Clausen, H.B.; Hammer, C.U.; Shoji, H. A 10-century comparison of prominent bipolar volcanic events in ice cores. J. Geophys. Res. Atmos. 1995, 100, 16241–16247. [Google Scholar] [CrossRef]
- Legrand, M.; Delmas, R.J. A 220-year continuous record of volcanic H2SO4 in the Antarctic ice sheet. Nature 1987, 327, 671–676. [Google Scholar] [CrossRef]
- Hattori, S.; Schmidt, J.A.; Johnson, M.S.; Danielache, S.O.; Yamada, A.; Ueno, Y.; Yoshida, N. SO2 photoexcitation mechanism links mass-independent sulfur isotopic fractionation in cryospheric sulfate to climate impacting volcanism. Proc. Natl. Acad. Sci. USA 2013, 110, 17656–17661. [Google Scholar] [CrossRef] [PubMed]
- Danielache, S.O.; Hattori, S.; Johnson, M.S.; Ueno, Y.; Nanbu, S.; Yoshida, N. Photoabsorption cross-section measurements of32S, 33S, 34S, and 36S sulfur dioxide for the B1B1-X1A1 absorption band. J. Geophys. Res. Atmos. 2012, 117, D24301. [Google Scholar] [CrossRef]
- Danielache, S.O.; Eskebjerg, C.; Johnson, M.S.; Ueno, Y.; Yoshida, N. High-precision spectroscopy of 32S, 33S, and 34S sulfur dioxide: Ultraviolet absorption cross sections and isotope effects. J. Geophys. Res. Atmos. 2008, 113, D17314. [Google Scholar] [CrossRef]
- Whitehill, A.R.; Jiang, B.; Guo, H.; Ono, S. SO2 photolysis as a source for sulfur mass-independent isotope signatures in stratospehric aerosols. Atmos. Chem. Phys. 2015, 15, 1843–1864. [Google Scholar] [CrossRef]
- Brasseur, G.P.; Solomon, S. Aeronomy of the Middle Atmosphere, Chemistry and Physics of the Stratosphere and Mesosphere; Springer: Dordrecht, The Netherlands, 2005. [Google Scholar]
- Bao, H.; Yu, S.; Tong, D.Q. Massive volcanic SO2 oxidation and sulphate aerosol deposition in Cenozoic North America. Nature 2010, 465, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Fraser, W.; Lomax, B.; Beerling, D.; James, D.; Pyle, J.; Self, S.; Sephton, M.; Wellman, C. Episodic perturbations of end-Permian atmosphere recorded in plant spore chemistry. In Proceedings of the EGU General Assembly 2016, Vienna, Austria, 17–22 April 2016; Volume 18, p. EPSC2016-17251. [Google Scholar]
- Lomax, B.H.; Fraser, W.T. Palaeoproxies: Botanical monitors and recorders of atmospheric change. Palaeontology 2015, 58, 759–768. [Google Scholar] [CrossRef]
- Shepherd, T.G. Issues in stratosphere-troposphere coupling. J. Meteorol. Soc. Jpn. 2002, 80, 769–792. [Google Scholar] [CrossRef]
- Robock, A. Climatic impact of volcanic emissions. In State of the Planet; Sparks, R.S.J., Hawkesworth, C.J., Eds.; American Geophysical Union: Washington, DC, USA, 2004; Volume 19. [Google Scholar]
- Bekki, S. Oxidation of Volcanic SO2: A Sink for Stratospheric OH and H2O. Geophys. Res. Lett. 1995, 22, 913–916. [Google Scholar] [CrossRef]




© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, E. Volcanic Plume Impact on the Atmosphere and Climate: O- and S-Isotope Insight into Sulfate Aerosol Formation. Geosciences 2018, 8, 198. https://doi.org/10.3390/geosciences8060198
Martin E. Volcanic Plume Impact on the Atmosphere and Climate: O- and S-Isotope Insight into Sulfate Aerosol Formation. Geosciences. 2018; 8(6):198. https://doi.org/10.3390/geosciences8060198
Chicago/Turabian StyleMartin, Erwan. 2018. "Volcanic Plume Impact on the Atmosphere and Climate: O- and S-Isotope Insight into Sulfate Aerosol Formation" Geosciences 8, no. 6: 198. https://doi.org/10.3390/geosciences8060198
APA StyleMartin, E. (2018). Volcanic Plume Impact on the Atmosphere and Climate: O- and S-Isotope Insight into Sulfate Aerosol Formation. Geosciences, 8(6), 198. https://doi.org/10.3390/geosciences8060198