Moringa Extract Attenuates Inflammatory Responses and Increases Gene Expression of Casein in Bovine Mammary Epithelial Cells


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Preparation of Moringa Extract (ME)
2.3. Determination of Free Radical Scavenging Activity
2.4. Cell Culture and Treatments
2.5. Cytotoxicity Test
2.6. Preparation of Cell Lysate, SDS-PAGE, and Western Blot Analysis
2.7. Nuclear Fractionation
2.8. Real-Time PCR Analysis
2.9. Assessment of Reactive Oxygen Species (ROS)
2.10. Statistical Analysis
3. Results
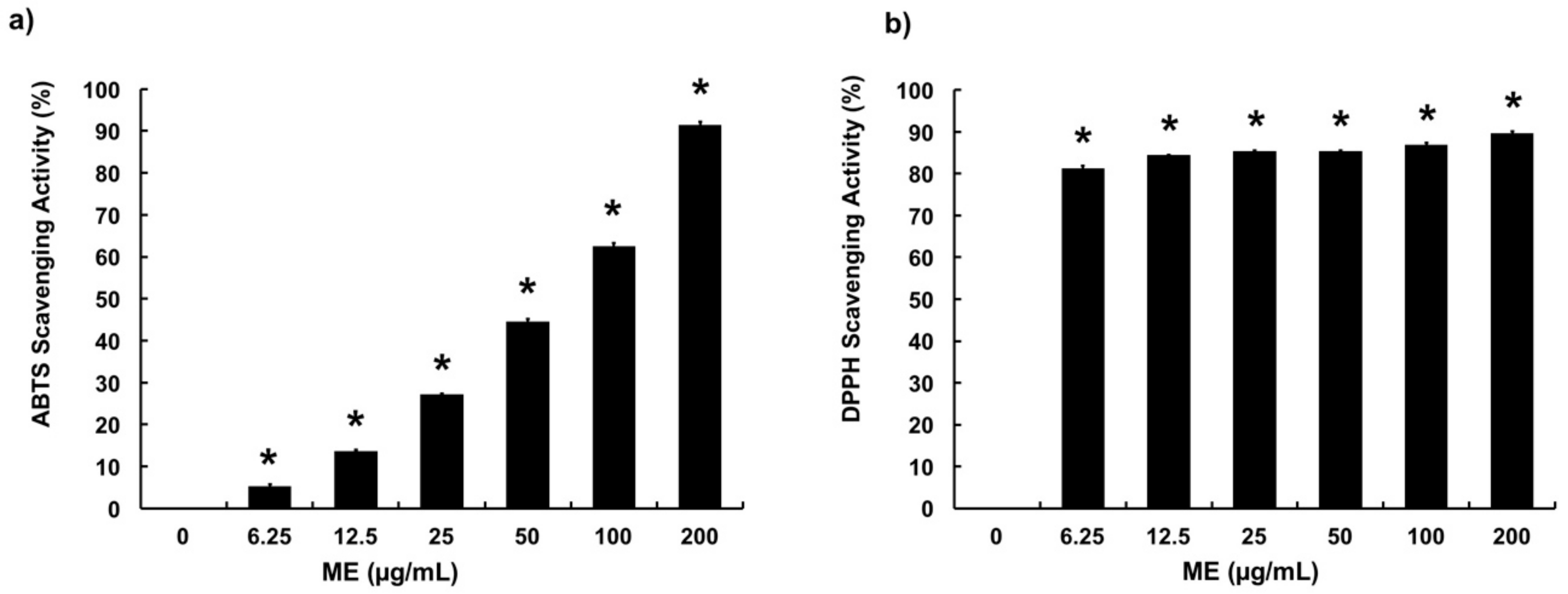
3.1. Free Radical Scavenging Activities of ME
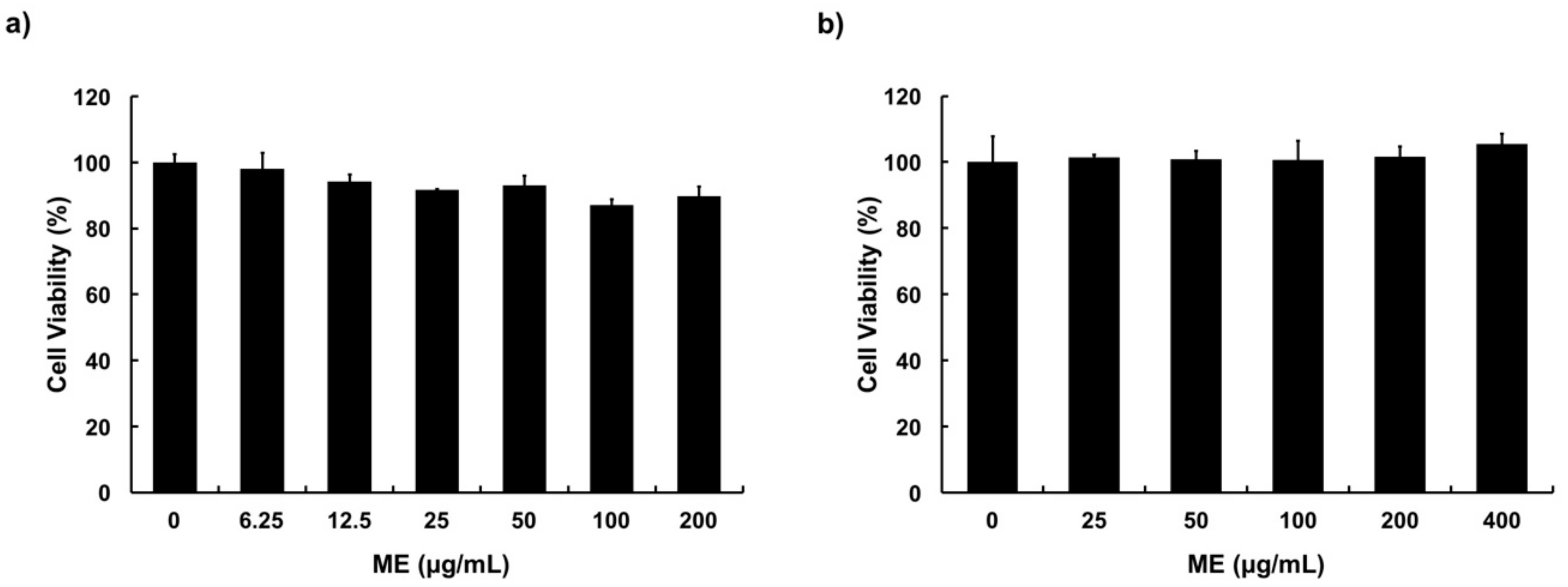
3.2. Cytotoxicity of ME in MAC-T Cells
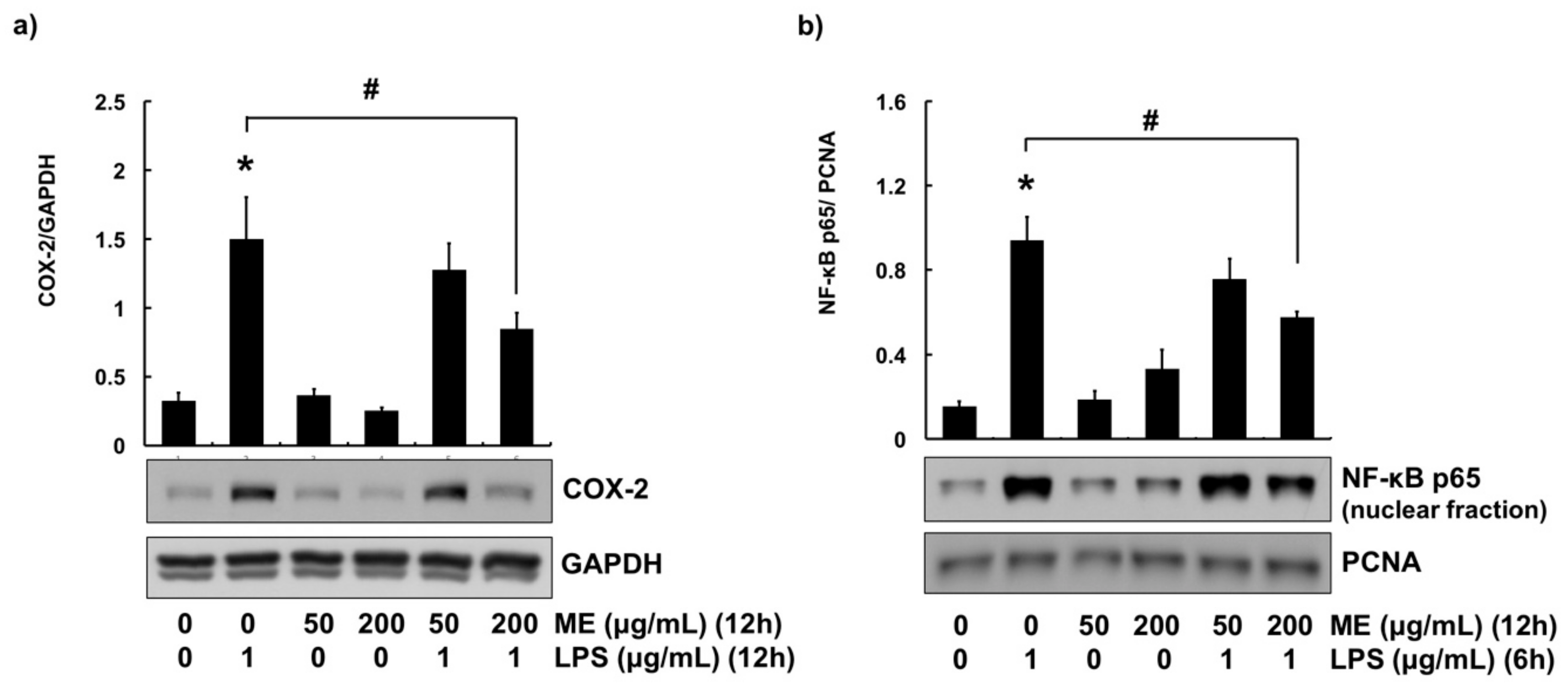
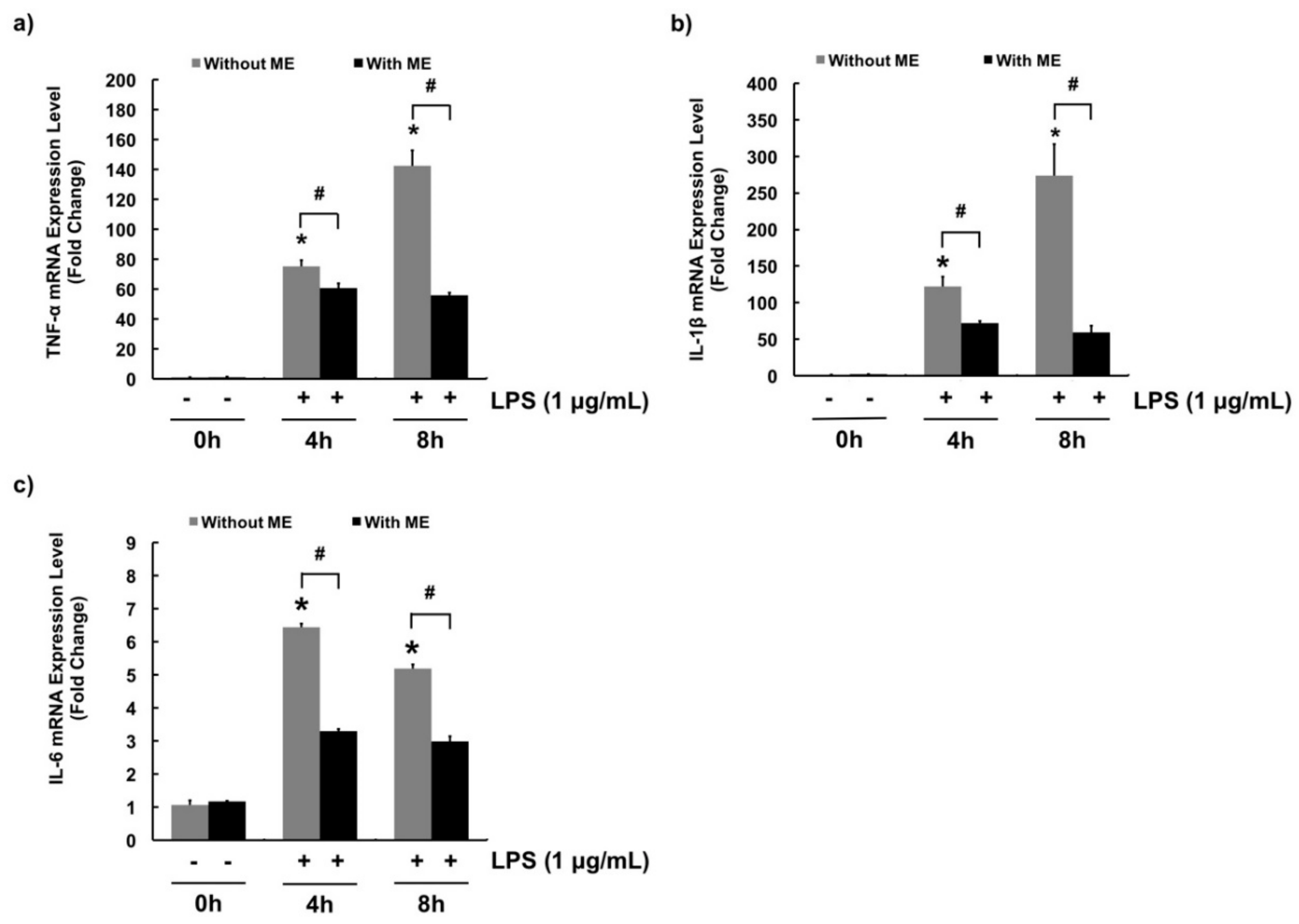
3.3. Anti-Inflammatory Effects of ME in MAC-T Cells
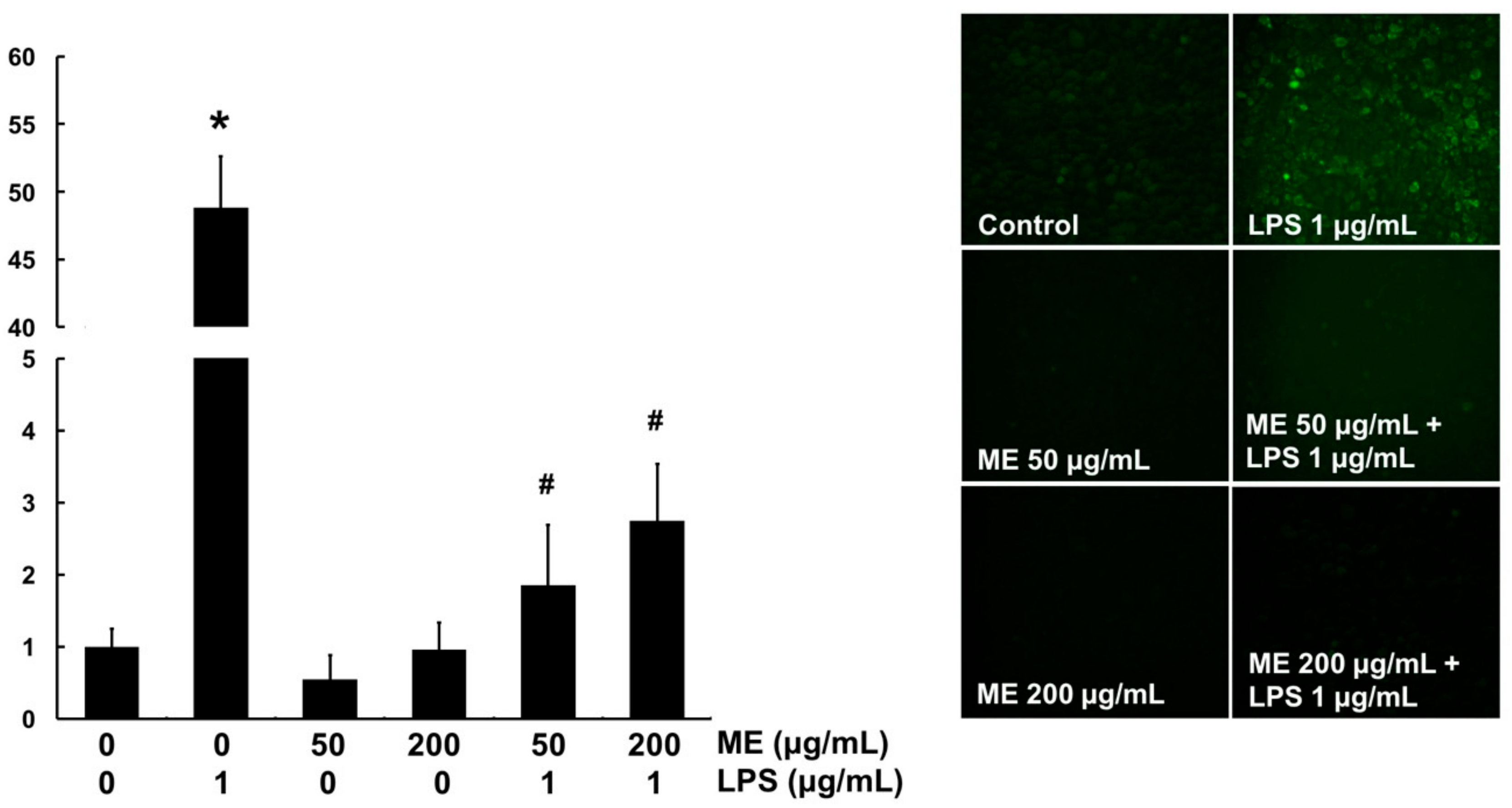
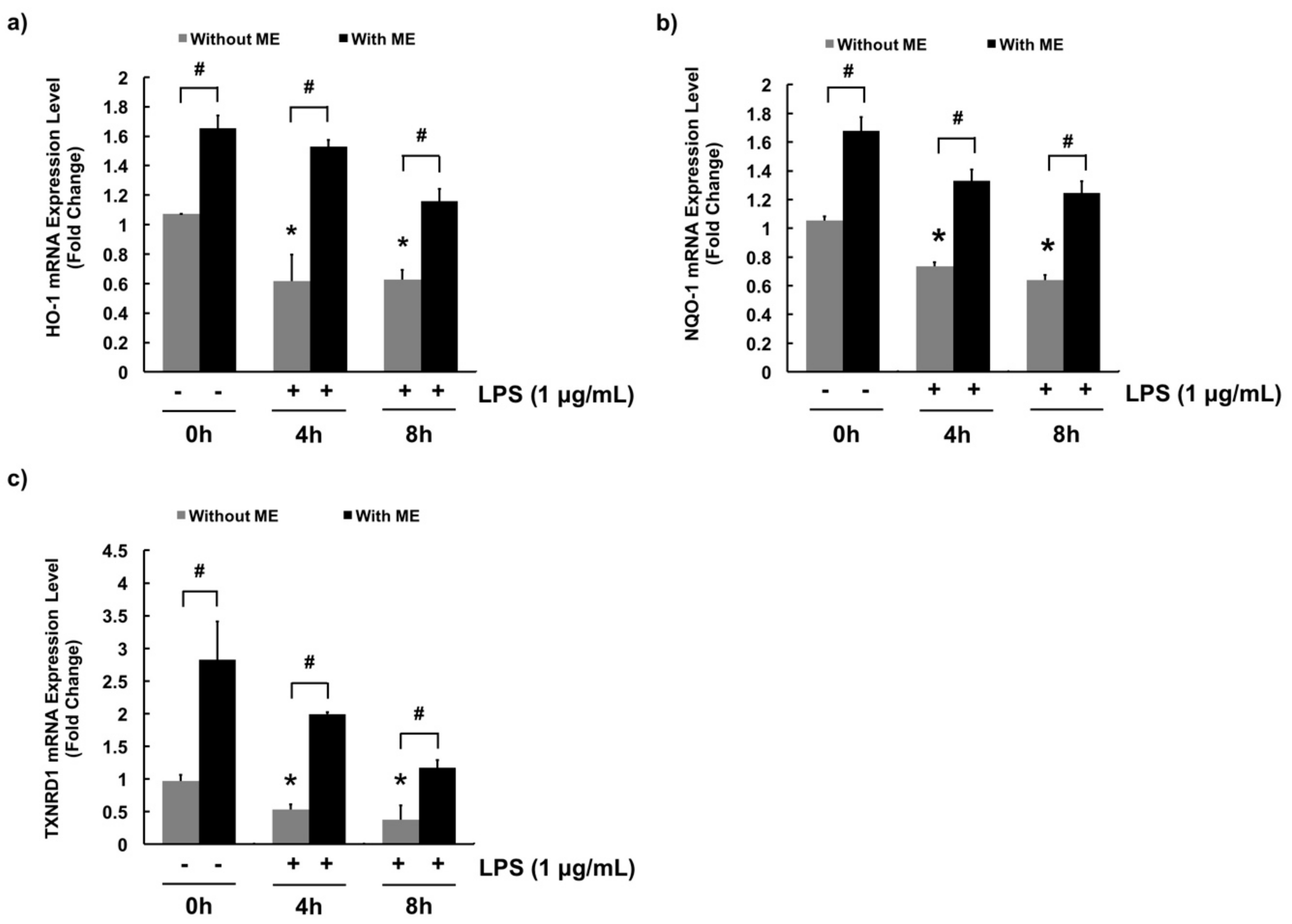
3.4. Antioxidant Effects of ME in MAC-T Cells
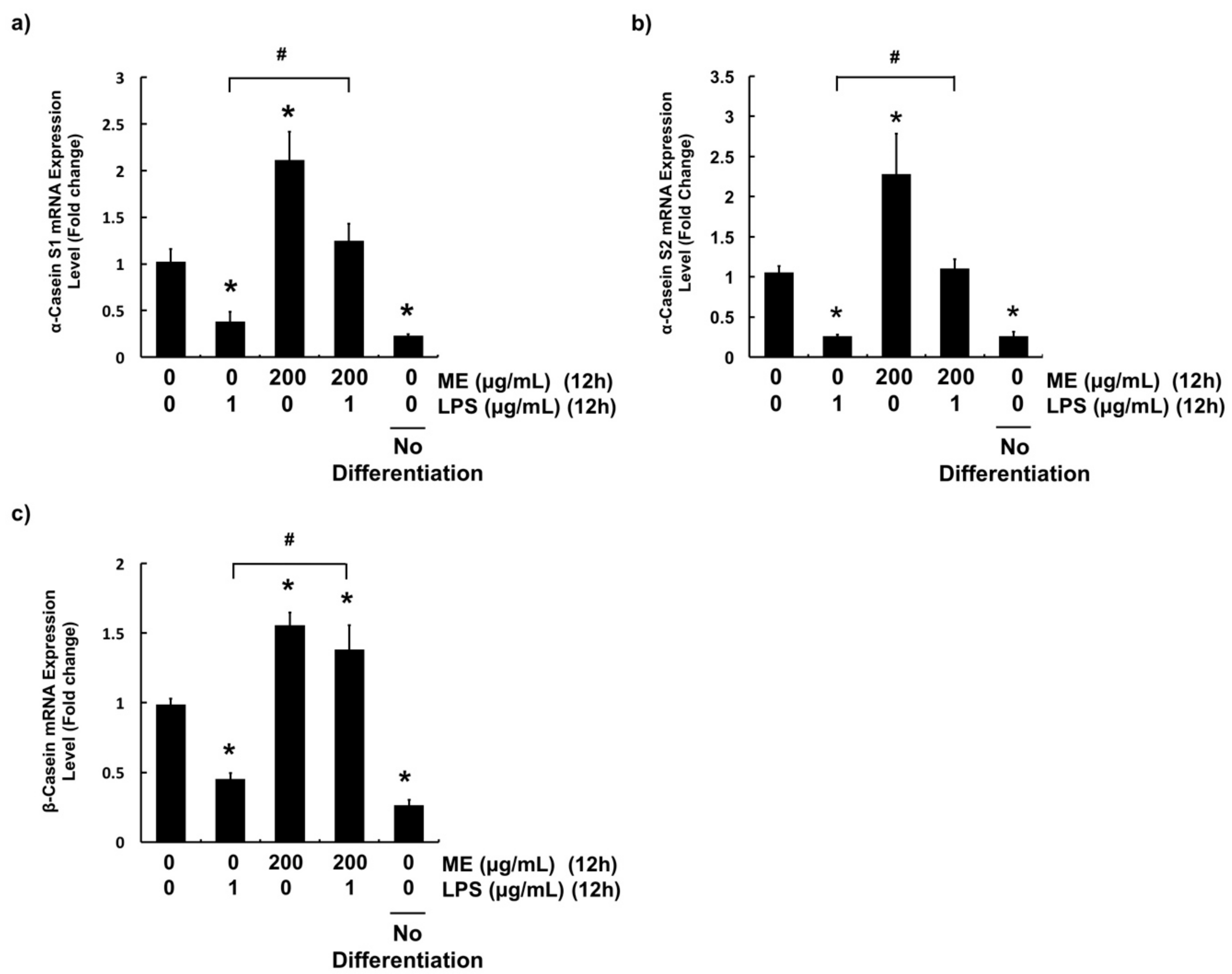
3.5. Effects of ME on Casein Production in Differentiated MAC-T Cells
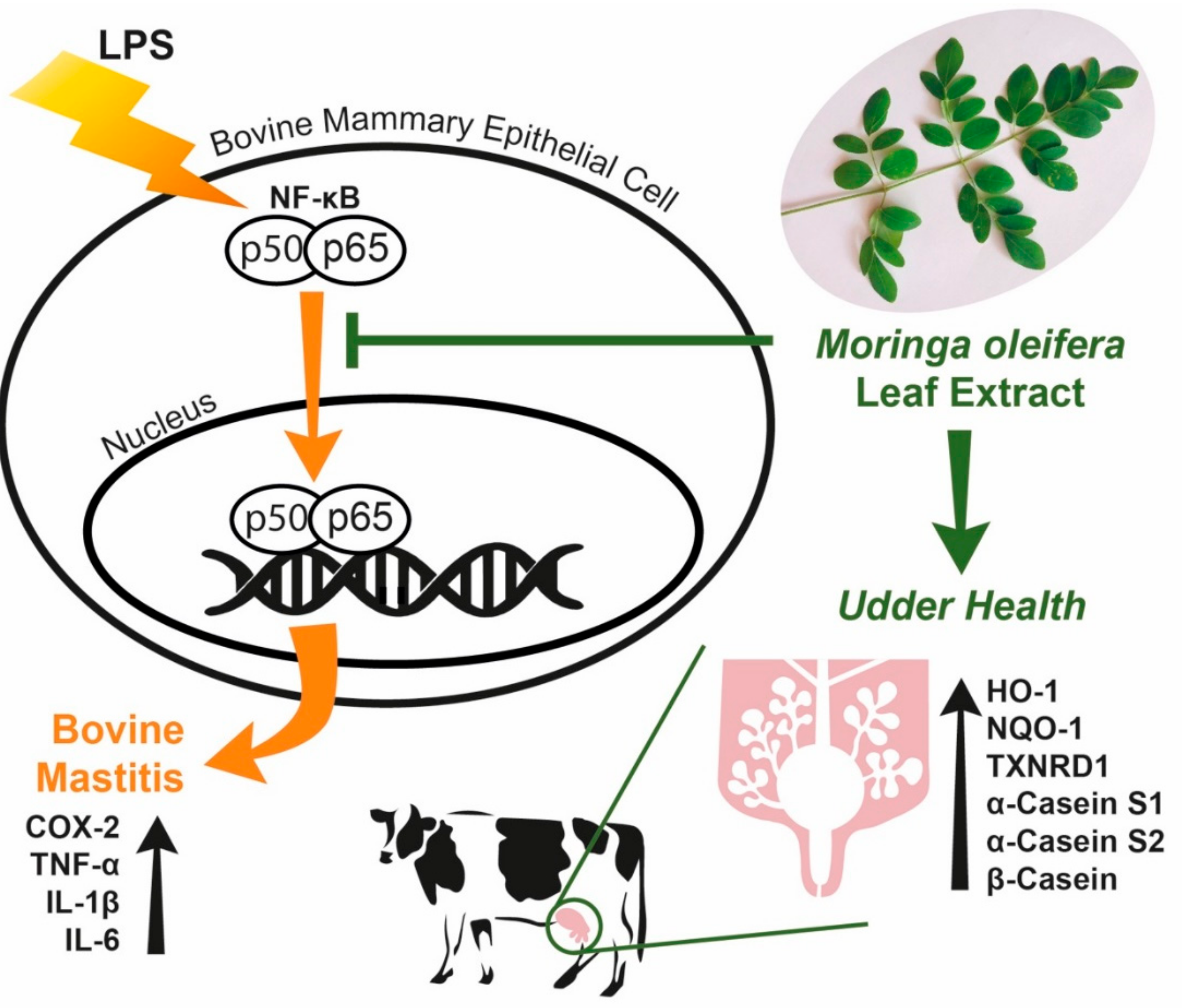
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Shaheen, M.; Tantary, H.; Nabi, S. A treatise on bovine mastitis: disease and disease economics, etiological basis, risk factors, impact on human health, therapeutic management, prevention and control strategy. J. Adv. Dairy Res. 2016, 4, 1–10. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Sig. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Jeong, C.H.; Cheng, W.N.; Bae, H.; Lee, K.W.; Han, S.M.; Petriello, M.C.; Lee, H.G.; Seo, H.G.; Han, S.G. Bee venom decreases LPS-induced inflammatory responses in bovine mammary epithelial cells. J. Microbiol. Biotechnol. 2017, 27, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Ur Rahman, S.; Zhang, L.; Shahid, M.; Zhang, S.; Liu, G.; Gao, J.; Han, B. ESBL-producing Escherichia coli from cows suffering mastitis in China contain clinical class 1 integrons with CTX-M linked to ISCR1. Front. Microbiol. 2016, 7, 1931. [Google Scholar] [CrossRef]
- Caudell, M.A.; Quinlan, M.B.; Quinlan, R.J.; Call, D.R. Medical pluralism and livestock health: ethnomedical and biomedical veterinary knowledge among East African agropastoralists. J. Ethnobiol. Ethnomed. 2017, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Zbinden, M.; Vogl, C.R.; Ivemeyer, S.; Meier, B.; Amorena, M.; Maeschli, A.; Hamburger, M.; Walkenhorst, M. Swiss ethnoveterinary knowledge on medicinal plants–a within-country comparison of Italian speaking regions with north-western German speaking regions. J. Ethnobiol. Ethnomed. 2017, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Vogl, C.R.; Amorena, M.; Hamburger, M.; Walkenhorst, M. Treatment of organic livestock with medicinal plants: a systematic review of European ethnoveterinary research. Complement. Med. Res. 2014, 21, 375–386. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wei, Z.; Zhou, E.; Chen, L.; Kou, J.; Wang, J.; Yang, Z. Baicalein attenuates inflammatory responses by suppressing TLR4 mediated NF-κB and MAPK signaling pathways in LPS-induced mastitis in mice. Int. Immunopharmacol. 2015, 28, 470–476. [Google Scholar] [CrossRef]
- Guo, M.; Zhang, N.; Li, D.; Liang, D.; Liu, Z.; Li, F.; Fu, Y.; Cao, Y.; Deng, X.; Yang, Z. Baicalin plays an anti-inflammatory role through reducing nuclear factor-κB and p38 phosphorylation in S. aureus-induced mastitis. Int. Immunopharmacol. 2013, 16, 125–130. [Google Scholar] [CrossRef]
- Wei, Z.; Zhou, E.; Guo, C.; Fu, Y.; Yu, Y.; Li, Y.; Yao, M.; Zhang, N.; Yang, Z. Thymol inhibits Staphylococcus aureus internalization into bovine mammary epithelial cells by inhibiting NF-κB activation. Microb. Pathog. 2014, 71, 15–19. [Google Scholar] [CrossRef]
- Suresh, S.; Sankar, P.; Telang, A.G.; Kesavan, M.; Sarkar, S.N. Nanocurcumin ameliorates Staphylococcus aureus-induced mastitis in mouse by suppressing NF‑κB signaling and inflammation. Int. Immunopharmocol. 2018, 65, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Gao, R.; Cao, Y.; Guo, M.; Wei, Z.; Zhou, E.; Li, Y.; Yao, M.; Yang, Z.; Zhang, N. Curcumin attenuates inflammatory responses by suppressing TLR4-mediated NF-κB signaling pathway in lipopolysaccharide-induced mastitis in mice. Int. Immunopharmacol. 2014, 20, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Zaku, S.; Emmanuel, S.; Tukur, A.; Kabir, A. Moringa oleifera: An underutilized tree in Nigeria with amazing versatility: A review. Afr. J. Food Sci. 2015, 9, 456–461. [Google Scholar]
- Abdull, R.; Ahmad, F.; Ibrahim, M.D.; Kntayya, S.B. Health benefits of Moringa oleifera. Asian Pac. J. Cancer Prev. 2014, 15, 8571–8576. [Google Scholar] [CrossRef] [PubMed]
- Ademiluyi, A.O.; Aladeselu, O.H.; Oboh, G.; Boligon, A.A. Drying alters the phenolic constituents, antioxidant properties, α-amylase, and α-glucosidase inhibitory properties of Moringa (Moringa oleifera) leaf. Food Sci. Nutr. 2018, 6, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, L.; Doriya, K.; Kumar, D.S. Moringa oleifera: A review on nutritive importance and its medicinal application. Food Sci. Hum. Wellness 2016, 5, 49–56. [Google Scholar] [CrossRef]
- Siddhuraju, P.; Becker, K. Antioxidant properties of various solvent extracts of total phenolic constituents from three different agroclimatic origins of drumstick tree (Moringa oleifera Lam.) leaves. J. Agric. Food Chem. 2003, 51, 2144–2155. [Google Scholar] [CrossRef]
- Shih, M.-C.; Chang, C.-M.; Kang, S.-M.; Tsai, M.-L. Effect of different parts (leaf, stem and stalk) and seasons (summer and winter) on the chemical compositions and antioxidant activity of Moringa oleifera. Int. J. Mol. Sci. 2011, 12, 6077–6088. [Google Scholar] [CrossRef]
- Zhang, T.; Jeong, C.H.; Cheng, W.N.; Bae, H.; Seo, H.G.; Petriello, M.C.; Han, S.G. Moringa extract enhances the fermentative, textural, and bioactive properties of yogurt. LWT. 2019, 101. [Google Scholar] [CrossRef]
- Kang, S.; Lee, J.S.; Lee, H.C.; Petriello, M.C.; Kim, B.Y.; Do, J.T.; Lim, D.-S.; Lee, H.G.; Han, S.G. Phytoncide extracted from pinecone decreases LPS-induced inflammatory responses in bovine mammary epithelial cells. J. Microbiol. Biotechnol. 2016, 26, 579–587. [Google Scholar] [CrossRef]
- Lee, H.; Heo, Y.; Lee, S.; Hwang, K.; Lee, H.; Choi, S.; Kim, N. Retinoic acid plus prolactin to synergistically increase specific casein gene expression in MAC-T cells. J. Dairy Sci. 2013, 96. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.T.; Ha, W.T.; Lee, R.; Lee, W.-Y.; Jeong, H.Y.; Hwang, K.C.; Song, H. Mammary alveolar cell as in vitro evaluation system for casein gene expression involved in glucose level. Asian-Australas. J. Anim. Sci. 2017, 30, 878. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.H.; Seok, J.S.; Petriello, M.C.; Han, S.G. Arsenic downregulates tight junction claudin proteins through p38 and NF-κB in intestinal epithelial cell line, HT-29. Toxicology 2017, 379. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Jeong, C.H.; Cheng, W.N.; Hong, K.; Seo, H.G.; Han, S.G. Oxidative stress-induced inflammatory responses and effects of N-acetylcysteine in bovine mammary alveolar cells. J. Dairy Res. 2017, 84, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, T.; Zhang, Z.; Jiang, H.; Wang, W.; Cao, Y.; Zhang, N. Leonurine exerts anti-inflammatory effect by regulating inflammatory signaling pathways and cytokines in LPS-induced mouse mastitis. Inflammation 2015, 38, 79–88. [Google Scholar] [CrossRef]
- Moyo, B.; Masika, P.J.; Hugo, A.; Muchenje, V. Nutritional characterization of Moringa (Moringa oleifera Lam.) leaves. Afr. J. Biotechnol. 2011, 10, 12925–12933. [Google Scholar]
- Jongrungruangchok, S.; Bunrathep, S.; Songsak, T. Nutrients and minerals content of eleven different samples of Moringa oleifera cultivated in Thailand. J. Health Res. 2010, 24, 123–127. [Google Scholar]
- Cohen-Zinder, M.; Weinberg, Z.; Leibovich, H.; Chen, Y.; Rosen, M.; Sagi, G.; Orlov, A.; Agmon, R.; Yishay, M.; Miron, J. Ensiled Moringa oleifera: an antioxidant-rich feed that improves dairy cattle performance. J. Agric. Sci. 2017, 155, 1174–1186. [Google Scholar] [CrossRef]
- Zeng, B.; Sun, J.; Chen, T.; Sun, B.; He, Q.; Chen, X.; Zhang, Y.; Xi, Q. Effects of Moringa oleifera silage on milk yield, nutrient digestibility and serum biochemical indexes of lactating dairy cows. J. Anim. Physiol. Anim. Nutr. 2018, 102, 75–81. [Google Scholar] [CrossRef]
- Mendieta-Araica, B.; Spörndly, E.; Reyes-Sánchez, N.; Spörndly, R. Feeding Moringa oleifera fresh or ensiled to dairy cows—effects on milk yield and milk flavor. Trop. Anim. Health Prod. 2011, 43, 1039–1047. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, C.; Quirantes-Piné, R.; Fernández-Gutiérrez, A.; Segura-Carretero, A. Optimization of extraction method to obtain a phenolic compounds-rich extract from Moringa oleifera Lam leaves. Ind. Crop. Prod. 2015, 66, 246–254. [Google Scholar] [CrossRef]
- Sankhalkar, S.; Vernekar, V. Quantitative and Qualitative analysis of Phenolic and Flavonoid content in Moringa oleifera Lam. and Ocimum tenuiflorum L. Pharmacognosy. Res. 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhen, J.; Villani, T.S.; Guo, Y.; Qi, Y.; Chin, K.; Pan, M.-H.; Ho, C.-T.; Simon, J.E.; Wu, Q. Phytochemistry, antioxidant capacity, total phenolic content and anti-inflammatory activity of Hibiscus sabdariffa leaves. Food Chem. 2016, 190, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Csepregi, K.; Neugart, S.; Schreiner, M.; Hideg, É. Comparative evaluation of total antioxidant capacities of plant polyphenols. Molecules 2016, 21, 208. [Google Scholar] [CrossRef] [PubMed]
- Fitriana, W.D.; Ersam, T.; Shimizu, K.; Fatmawati, S. Antioxidant activity of Moringa oleifera extracts. Indones. J. Chem. 2016, 16, 297–301. [Google Scholar] [CrossRef]
- Floegel, A.; Kim, D.-O.; Chung, S.-J.; Koo, S.I.; Chun, O.K. Comparison of ABTS/DPPH assays to measure antioxidant capacity in popular antioxidant-rich US foods. J. Food Compost. Anal. 2011, 24, 1043–1048. [Google Scholar] [CrossRef]
- Kim, D.-O.; Lee, K.W.; Lee, H.J.; Lee, C.Y. Vitamin C equivalent antioxidant capacity (VCEAC) of phenolic phytochemicals. J. Agric. Food Chem. 2002, 50, 3713–3717. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.T.; Robitaille, G.; Turner, J.D. Establishment of bovine mammary epithelial cells (MAC-T): An in vitro model for bovine lactation. Exp. Cell Res. 1991, 197, 191–199. [Google Scholar] [CrossRef]
- Günther, J.; Koy, M.; Berthold, A.; Schuberth, H.-J.; Seyfert, H.-M. Comparison of the pathogen species-specific immune response in udder derived cell types and their models. Vet. Res. 2016, 47, 22. [Google Scholar] [CrossRef]
- Gandhi, J.; Khera, L.; Gaur, N.; Paul, C.; Kaul, R. Role of modulator of inflammation cyclooxygenase-2 in gammaherpesvirus mediated tumorigenesis. Front. Microbiol. 2017, 8, 538. [Google Scholar] [CrossRef] [PubMed]
- Fard, M.T.; Arulselvan, P.; Karthivashan, G.; Adam, S.K.; Fakurazi, S. Bioactive extract from Moringa oleifera inhibits the pro-inflammatory mediators in lipopolysaccharide stimulated macrophages. Pharmacogn. Mag. 2015, 11, 556–563. [Google Scholar]
- Arulselvan, P.; Tan, W.; Gothai, S.; Muniandy, K.; Fakurazi, S.; Esa, N.; Alarfaj, A.; Kumar, S. Anti-inflammatory potential of ethyl acetate fraction of Moringa oleifera in downregulating the NF-κB signaling pathway in lipopolysaccharide-stimulated macrophages. Molecules 2016, 21, 1452. [Google Scholar] [CrossRef] [PubMed]
- Azizah, D.; Masfria, K.; Poppy, A. Ethanol Extract and active fraction effect of Moringa oleifera. Lam in inhibiting COX-2 activity on MCF-7 cell. Asian J. Pharm. Res. Dev. 2018, 6. [Google Scholar]
- Shi, H.; Yan, S.; Guo, Y.; Zhang, B.; Guo, X.; Shi, B. Vitamin A pretreatment protects NO-induced bovine mammary epithelial cells from oxidative stress by modulating Nrf2 and NF-κB signaling pathways. J. Anim. Sci. 2018, 96, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Ezzat Alnakip, M.; Quintela-Baluja, M.; Böhme, K.; Fernández-No, I.; Caamaño-Antelo, S.; Calo-Mata, P.; Barros-Velázquez, J. The immunology of mammary gland of dairy ruminants between healthy and inflammatory conditions. J. Vet. Med. 2014, 2014, 659801. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Dejie, L.; Xiaojing, S.; Tiancheng, W.; Yongguo, C.; Zhengtao, Y.; Naisheng, Z. Magnolol inhibits the inflammatory response in mouse mammary epithelial cells and a mouse mastitis model. Inflammation 2015, 38, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Wang, K.; Liu, H.; Hu, F.; Zhao, F.; Liu, J. Protection of bovine mammary epithelial cells from hydrogen peroxide-induced oxidative cell damage by resveratrol. Oxid. Med. Cell. Longev. 2016, 2016, 2572175. [Google Scholar] [CrossRef] [PubMed]
- Adedapo, A.A.; Falayi, O.O.; Oyagbemi, A.A. Evaluation of the analgesic, anti-inflammatory, anti-oxidant, phytochemical and toxicological properties of the methanolic leaf extract of commercially processed Moringa oleifera in some laboratory animals. J. Basic Clin. Physiol. Pharmacol. 2015, 26. [Google Scholar] [CrossRef] [PubMed]
- Abdou, K.; Moselhy, W.A.; Mohamed, H.M.; El-Nahass, E.-S.; Khalifa, A.G. Moringa oleifera Leaves Extract Protects Titanium Dioxide Nanoparticles-Induced Nephrotoxicity via Nrf2/HO-1 Signaling and Amelioration of Oxidative Stress. Biol. Trace Elem. Res. 2019, 187, 181–191. [Google Scholar] [CrossRef]
- Arulselvan, P.; Fard, M.T.; Tan, W.S.; Gothai, S.; Fakurazi, S.; Norhaizan, M.E.; Kumar, S.S. Role of antioxidants and natural products in inflammation. Oxid. Med. Cell. Longev. 2016, 2016, 5276130. [Google Scholar] [CrossRef]
- Charoensin, S. Antioxidant and anticancer activities of Moringa oleifera leaves. J. Med. Plants Res. 2014, 8, 318–325. [Google Scholar]
- Jaja-Chimedza, A.; Zhang, L.; Wolff, K.; Graf, B.L.; Kuhn, P.; Moskal, K.; Carmouche, R.; Newman, S.; Salbaum, J.M.; Raskin, I. A dietary isothiocyanate-enriched moringa (Moringa oleifera) seed extract improves glucose tolerance in a high-fat-diet mouse model and modulates the gut microbiome. J. Funct. Foods 2018, 47, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Omodanisi, E.; Aboua, Y.; Oguntibeju, O. Assessment of the anti-hyperglycaemic, anti-inflammatory and antioxidant activities of the methanol extract of Moringa oleifera in diabetes-induced nephrotoxic male wistar rats. Molecules 2017, 22, 439. [Google Scholar] [CrossRef] [PubMed]
- Vergara-Jimenez, M.; Almatrafi, M.; Fernandez, M. Bioactive components in Moringa oleifera leaves protect against chronic disease. Antioxidants 2017, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.; Li, B.; Olayanju, J.; Drake, J.; Chen, N. Nutraceutical or Pharmacological Potential of Moringa oleifera Lam. Nutrients 2018, 10, 343. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Young, J.; Chanson, P.; Binart, N. New insights in prolactin: pathological implications. Nat. Rev. Endocrinol. 2015, 11, 265. [Google Scholar] [CrossRef] [PubMed]
- Binart, N. Prolactin. In The Pituitary, 4th ed.; Melmed, S., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 129–161. [Google Scholar]
- Feuermann, Y.; Mabjeesh, S.J.; Shamay, A. Mammary fat can adjust prolactin effect on mammary epithelial cells via leptin and estrogen. Int. J. Endocrinol. 2009, 2009, 659801. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, K.A.; Shea, M.P.; Salituro, S.; Blohm, C.E.; Schuler, L.A. Prolactin alters the mammary epithelial hierarchy, increasing progenitors and facilitating ovarian steroid action. Stem Cell Rep. 2017, 9, 1167–1179. [Google Scholar] [CrossRef]
- Lee, W.-Y.; Park, H.-J.; Yeo, J.M.; Jeong, H.Y.; Song, H. Enhancement of milk protein expression in mammary epithelial cells via co-culturing with preadipocyte cells. Biotechnol. Bioprocess Eng. 2017, 22, 556–560. [Google Scholar] [CrossRef]
- Hennighausen, L.; Robinson, G.W.; Wagner, K.-U.; Liu, X. Prolactin signaling in mammary gland development. J. Biol. Chem. 1997, 272, 7567–7569. [Google Scholar] [CrossRef]
- Wu, T.; Wang, C.; Ding, L.; Shen, Y.; Cui, H.; Wang, M.; Wang, H. Arginine relieves the inflammatory response and enhances the casein expression in bovine mammary epithelial cells induced by lipopolysaccharide. Mediators Inflamm. 2016, 2016, 9618795. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, B.; Wang, H.; Bu, D.; Wang, J.; Loor, J.-J. Effects of arginine concentration on the in vitro expression of casein and mTOR pathway related genes in mammary epithelial cells from dairy cattle. PLoS ONE 2014, 9, e95985. [Google Scholar] [CrossRef] [PubMed]
- Titi, M.; Harijono, E.; Endang, S. Effect lactagogue moringa leaves (Moringa oleifera Lam.) powder in rats white female wistar. J. Basic Appl. Sci. Res. 2013, 3, 430–434. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence 5’-3’ |
|---|---|
| GAPDH | (F) ATG ATT CCA CCC ACG GCA AGT T (R) ACC ACA TAC TCA GCA CCA GCA T |
| TNF-α | (F) ACG GGC TTT ACC TCA TCT ACT CAC (R) TTG ACC TTG GTC TGG TAG GAG ACT |
| IL-1β | (F) CCG TAC CTG AAC CCA TCA ACG AAA (R) GGT GTT GGA TGC AGC TCT TCA TCT |
| IL-6 | (F) AGC GCA TGG TCG ACA AAA TCT C (R) AAC CCA GAT TGG AAG CAT CCG T |
| HO-1 | (F) AGG ATT TGT CAG AGG CCC TGA A (R) CAA AGA CGC CAT CAC CAG CTT A |
| NQO-1 | (F) GGT GCT CAT AGG GGA GTT CG (R) GGG AGT GTG CCC AAT GCT AT |
| TXNRD1 | (F) CGG TAT TGC TGG CAA TAG GAA GAG (R) GGC ATA GAT GTA AGG CAC GTT GGT |
| α-casein S1 | (F) GGG AAT CCAT GCC CAA CAG AAA GA (R) GGA ACG TAA TAC CAG GCA CCA GAT |
| α-casein S2 | (F) GGA CGA TAA GCA CTA CCA GAA AGC (R) AGA GTG GGA GTA ATG GGA ACA GCA |
| β-casein | (F) CCT AAC AGC CTC CCA CAA AA (R) AGA CTG GAG CAG AGG CAG AG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.N.; Jeong, C.H.; Seo, H.G.; Han, S.G. Moringa Extract Attenuates Inflammatory Responses and Increases Gene Expression of Casein in Bovine Mammary Epithelial Cells. Animals 2019, 9, 391. https://doi.org/10.3390/ani9070391
Cheng WN, Jeong CH, Seo HG, Han SG. Moringa Extract Attenuates Inflammatory Responses and Increases Gene Expression of Casein in Bovine Mammary Epithelial Cells. Animals. 2019; 9(7):391. https://doi.org/10.3390/ani9070391
Chicago/Turabian StyleCheng, Wei Nee, Chang Hee Jeong, Han Geuk Seo, and Sung Gu Han. 2019. "Moringa Extract Attenuates Inflammatory Responses and Increases Gene Expression of Casein in Bovine Mammary Epithelial Cells" Animals 9, no. 7: 391. https://doi.org/10.3390/ani9070391
APA StyleCheng, W. N., Jeong, C. H., Seo, H. G., & Han, S. G. (2019). Moringa Extract Attenuates Inflammatory Responses and Increases Gene Expression of Casein in Bovine Mammary Epithelial Cells. Animals, 9(7), 391. https://doi.org/10.3390/ani9070391