
Genomic Analysis of Indel and SV Reveals Functional and Adaptive Signatures in Hubei Indigenous Cattle Breeds
 , and
, and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
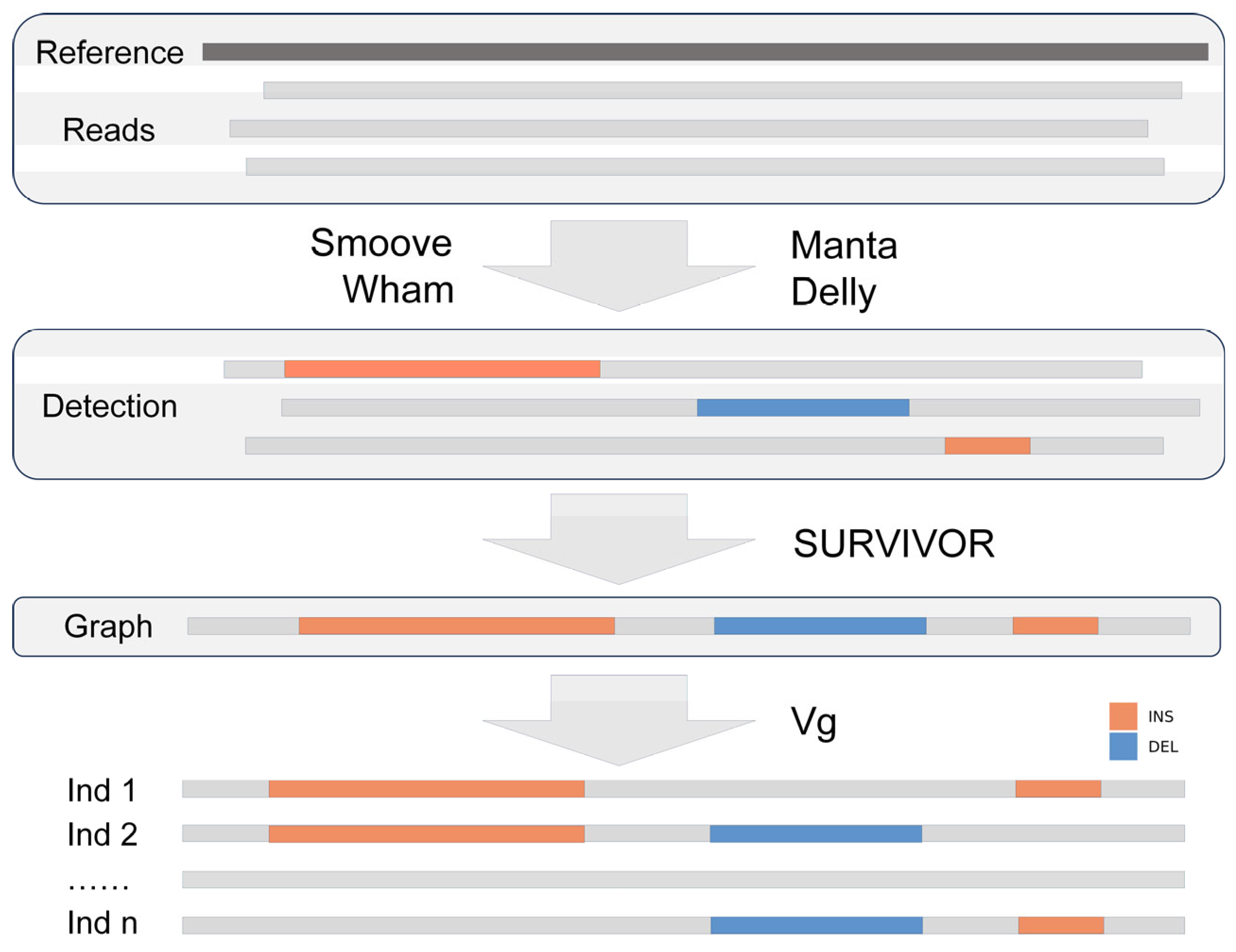
2.1. Sample Collection, Genomic Resequencing Read Filtering and Alignment
2.2. Variant Calling and Filtering
2.3. Identification of Insertions and Deletions Hotspots
2.4. Identification of Genomic Repetitive Sequences in Hubei Cattle
2.5. Functional Annotation of Deletions and Insertions in Regulatory and Functional Genomic Regions
2.6. Functional Annotation of Indels and SVs in Regulatory and Functional Genomic Regions
2.7. Population Structure Analysis
2.8. Annotation and Enrichment Analysis of Indels and SVs
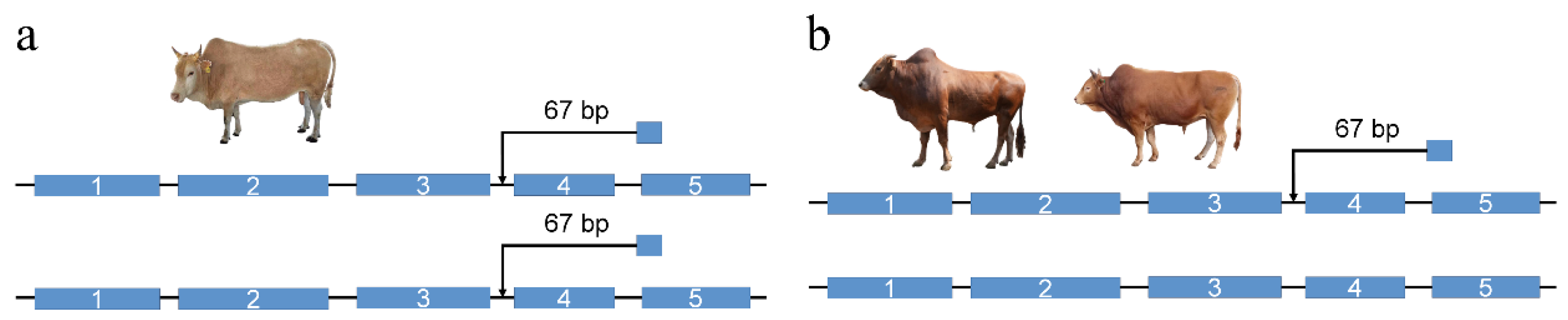
2.9. PCR Validation of the NOTCH2 67 bp Insertion
3. Results
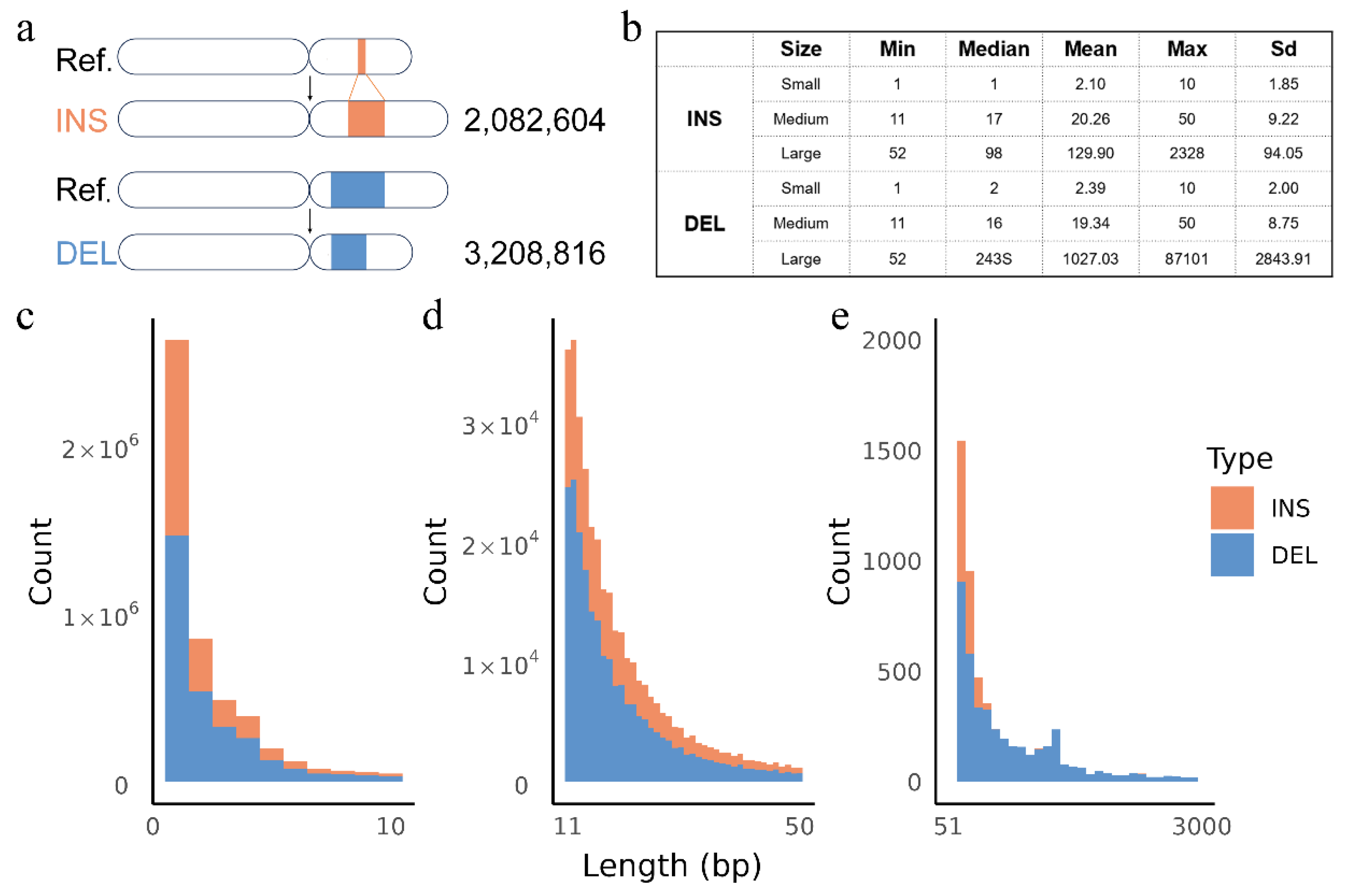
3.1. Overview of Resequencing Data and Identified Variants in Hubei Indigenous Cattle
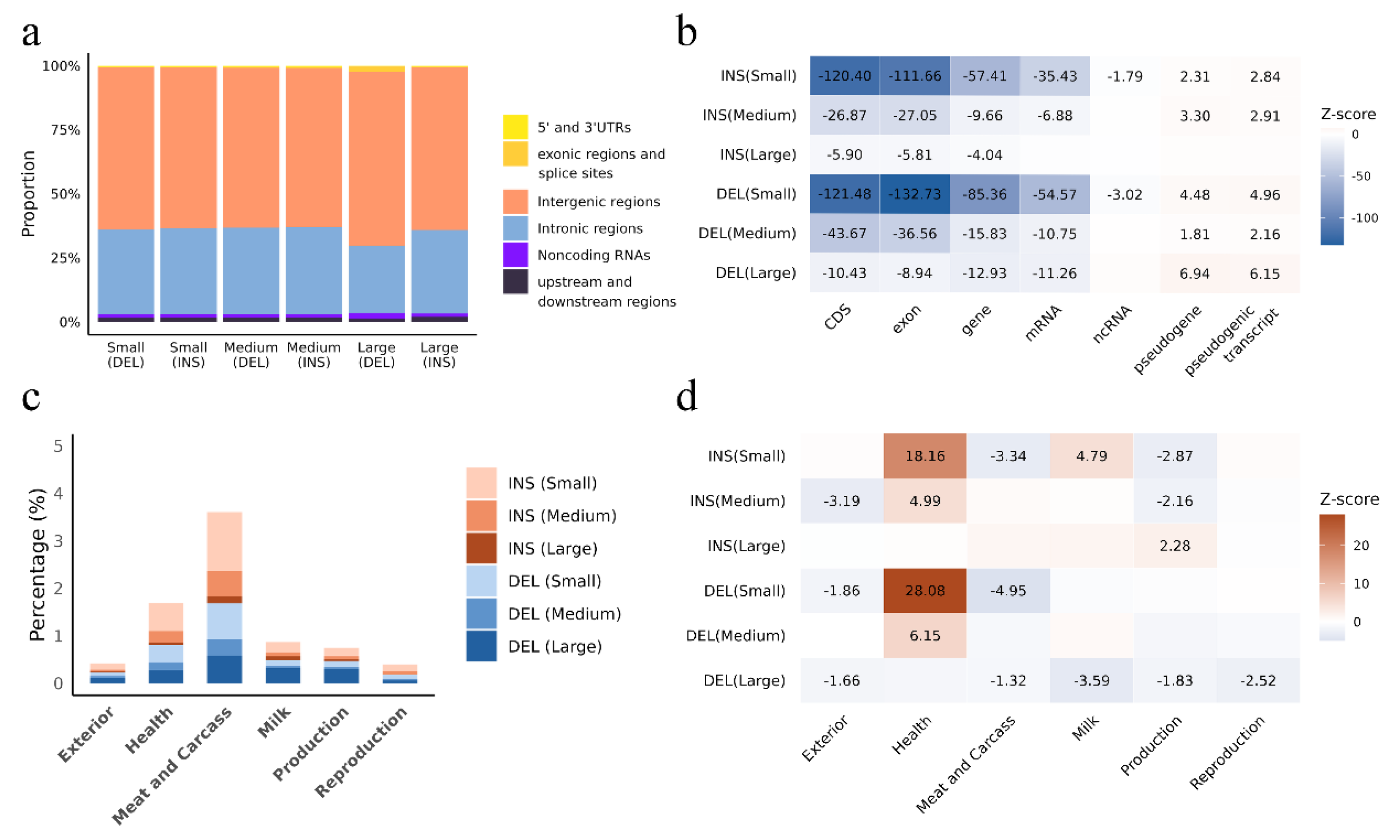
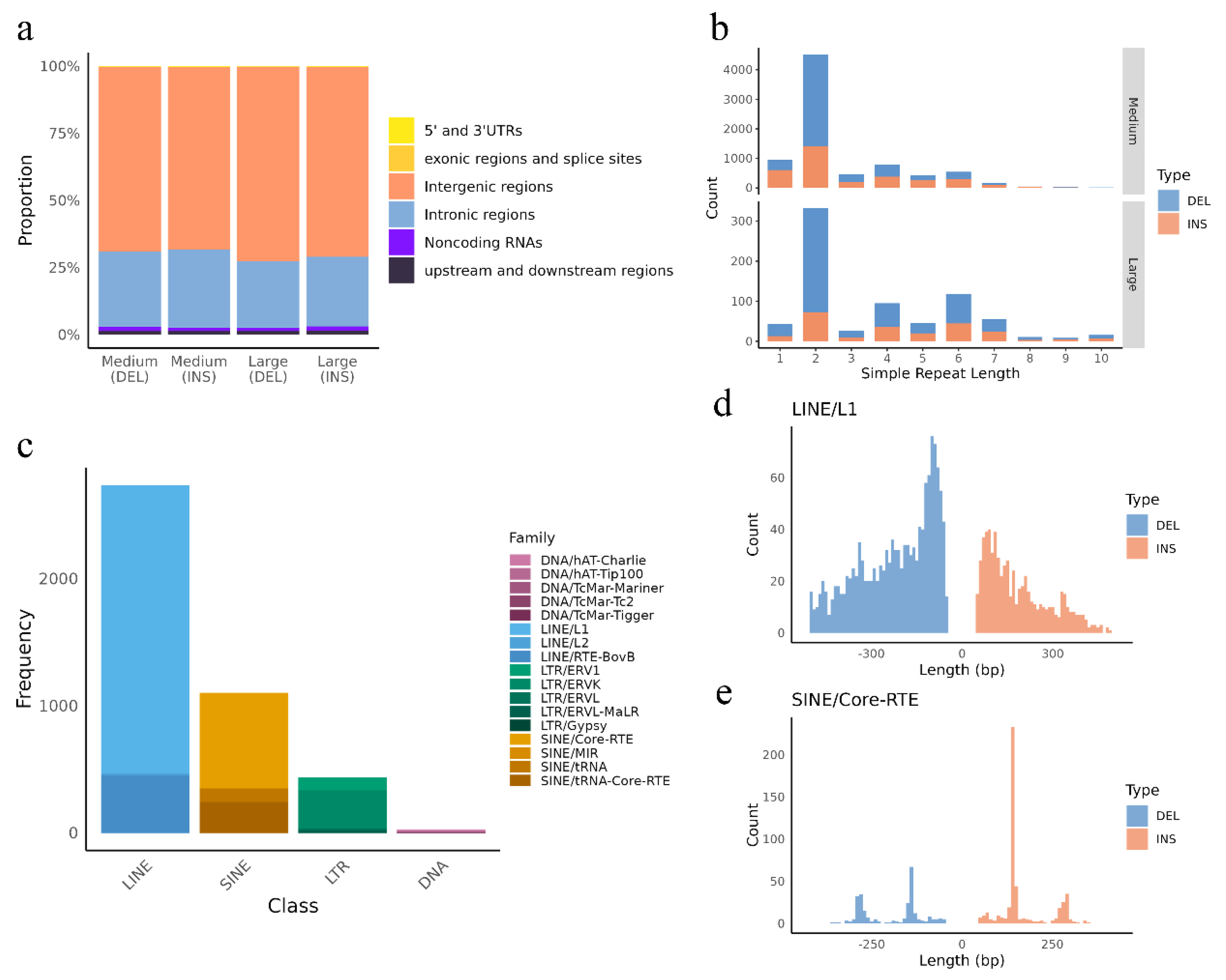
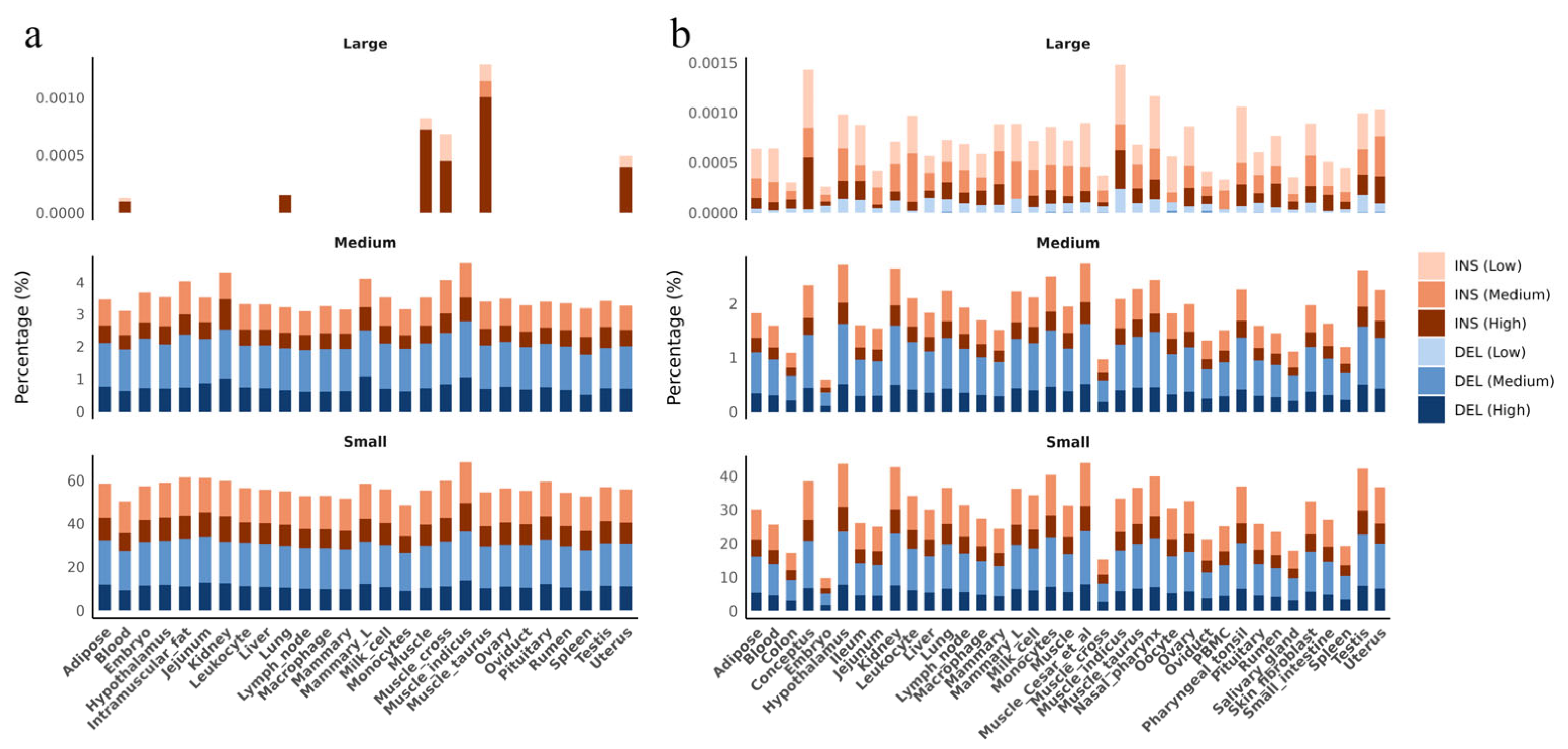
3.2. Insertions and Deletions Overlap with Genes, Regulatory Elements and QTLs
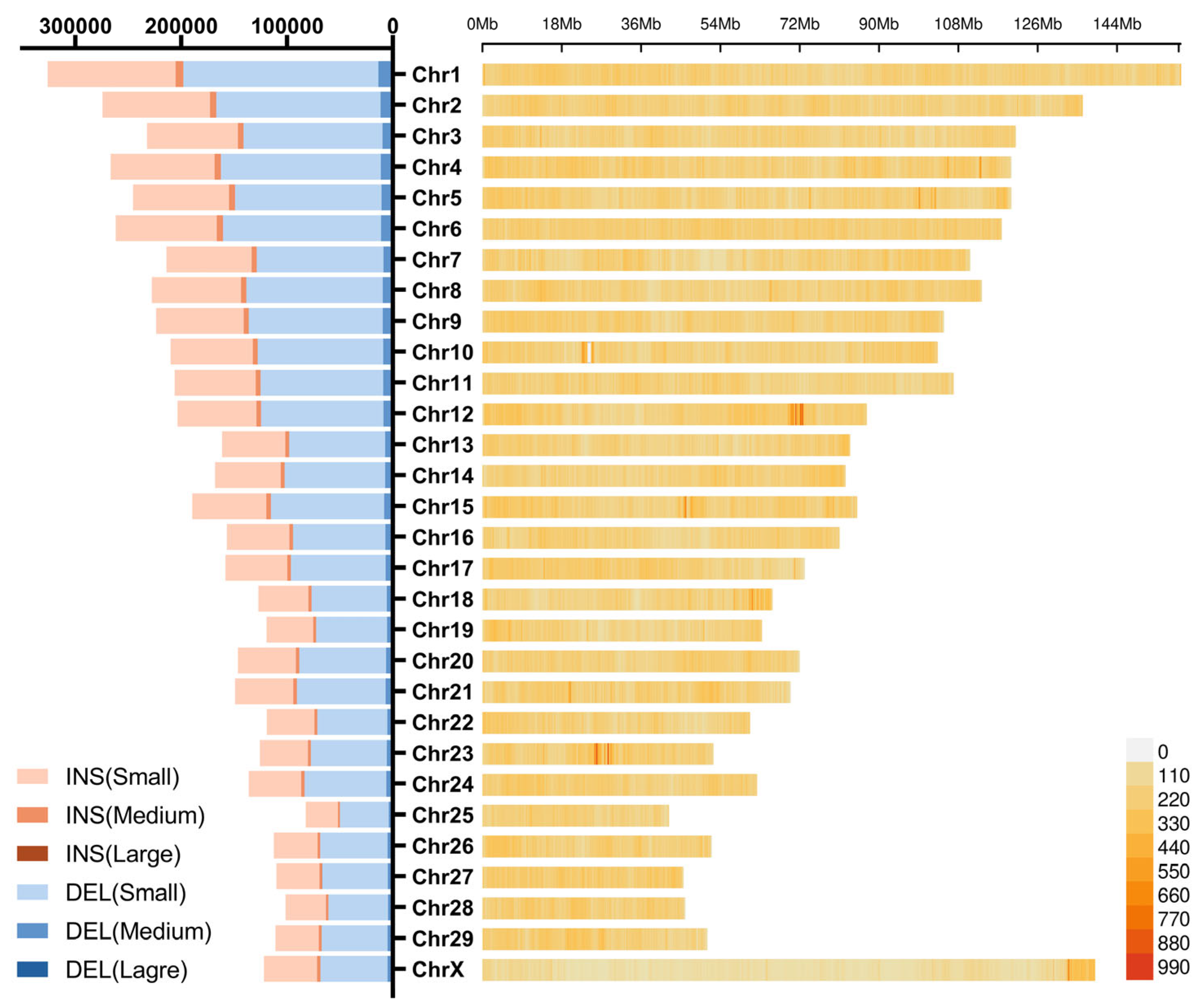
3.3. Distribution of Insertions and Deletions Hotspots
3.4. Repeat-Driven DEL and INSs
3.5. LD-Tag
3.6. Population Genetic Differentiation Based on Fst Analysis
3.7. NOTCH2 Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilbert, M.; Nicolas, G.; Cinardi, G.; Van Boeckel, T.P.; Vanwambeke, S.O.; Wint, G.R.W.; Robinson, T.P. Global distribution data for cattle, buffaloes, horses, sheep, goats, pigs, chickens and ducks in 2010. Sci. Data 2018, 5, 180227. [Google Scholar] [CrossRef] [PubMed]
- Latawiec, A.E.; Strassburg, B.B.; Valentim, J.F.; Ramos, F.; Alves-Pinto, H.N. Intensification of cattle ranching production systems: Socioeconomic and environmental synergies and risks in Brazil. Animal 2014, 8, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kwon, T.; Dessie, T.; Yoo, D.; Mwai, O.A.; Jang, J.; Sung, S.; Lee, S.; Salim, B.; Jung, J.; et al. The mosaic genome of indigenous African cattle as a unique genetic resource for African pastoralism. Nat. Genet. 2020, 52, 1099–1110. [Google Scholar] [CrossRef]
- Guan, X.; Xiang, W.; Qu, K.; Ahmed, Z.; Liu, J.; Cai, M.; Zhang, J.; Chen, N.; Lei, C.; Huang, B. Whole genome insights into genetic diversity, introgression, and adaptation of Yunnan indigenous cattle of Southwestern China. BMC Genom. 2025, 26, 216. [Google Scholar] [CrossRef]
- Buggiotti, L.; Yurchenko, A.A.; Yudin, N.S.; Vander Jagt, C.J.; Vorobieva, N.V.; Kusliy, M.A.; Vasiliev, S.K.; Rodionov, A.N.; Boronetskaya, O.I.; Zinovieva, N.A.; et al. Demographic History, Adaptation, and NRAP Convergent Evolution at Amino Acid Residue 100 in the World Northernmost Cattle from Siberia. Mol. Biol. Evol. 2021, 38, 3093–3110. [Google Scholar] [CrossRef]
- Gualdron Duarte, J.L.; Yuan, C.; Gori, A.S.; Moreira, G.C.M.; Takeda, H.; Coppieters, W.; Charlier, C.; Georges, M.; Druet, T. Sequenced-based GWAS for linear classification traits in Belgian Blue beef cattle reveals new coding variants in genes regulating body size in mammals. Genet. Sel. Evol. 2023, 55, 83. [Google Scholar] [CrossRef]
- Niu, Q.; Zhang, T.; Xu, L.; Wang, T.; Wang, Z.; Zhu, B.; Zhang, L.; Gao, H.; Song, J.; Li, J.; et al. Integration of selection signatures and multi-trait GWAS reveals polygenic genetic architecture of carcass traits in beef cattle. Genomics 2021, 113, 3325–3336. [Google Scholar] [CrossRef]
- Sanchez, M.P.; Tribout, T.; Kadri, N.K.; Chitneedi, P.K.; Maak, S.; Hoze, C.; Boussaha, M.; Croiseau, P.; Philippe, R.; Spengeler, M.; et al. Sequence-based GWAS meta-analyses for beef production traits. Genet. Sel. Evol. 2023, 55, 70. [Google Scholar] [CrossRef]
- Fonseca, P.A.S.; Caldwell, T.; Mandell, I.; Wood, K.; Canovas, A. Genome-wide association study for meat tenderness in beef cattle identifies patterns of the genetic contribution in different post-mortem stages. Meat Sci. 2022, 186, 108733. [Google Scholar] [CrossRef]
- Arikawa, L.M.; Mota, L.F.M.; Schmidt, P.I.; Frezarim, G.B.; Fonseca, L.F.S.; Magalhaes, A.F.B.; Silva, D.A.; Carvalheiro, R.; Chardulo, L.A.L.; Albuquerque, L.G. Genome-wide scans identify biological and metabolic pathways regulating carcass and meat quality traits in beef cattle. Meat Sci. 2024, 209, 109402. [Google Scholar] [CrossRef]
- Twomey, A.J.; Berry, D.P.; Evans, R.D.; Doherty, M.L.; Graham, D.A.; Purfield, D.C. Genome-wide association study of endo-parasite phenotypes using imputed whole-genome sequence data in dairy and beef cattle. Genet. Sel. Evol. 2019, 51, 15. [Google Scholar] [CrossRef] [PubMed]
- Recuerda, M.; Campagna, L. How structural variants shape avian phenotypes: Lessons from model systems. Mol. Ecol. 2024, 33, e17364. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Zhao, Y.; Zhu, L.; Li, X.; Zhang, J.; Cui, X.; Li, W.; Hao, D.; Yang, Z.; Wu, F.; et al. Genetic dissection of ten photosynthesis-related traits based on InDel- and SNP-GWAS in soybean. Theor. Appl. Genet. 2024, 137, 96. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, M.; Guo, Z.; Wijayanti, D.; Xu, H.; Jiang, F.; Lan, X. Insertion/Deletion (InDel) Variants within the Sheep Fat-Deposition-Related PDGFD Gene Strongly Affect Morphological Traits. Animals 2023, 13, 1485. [Google Scholar] [CrossRef]
- Das, S.; Upadhyaya, H.D.; Srivastava, R.; Bajaj, D.; Gowda, C.L.; Sharma, S.; Singh, S.; Tyagi, A.K.; Parida, S.K. Genome-wide insertion-deletion (InDel) marker discovery and genotyping for genomics-assisted breeding applications in chickpea. DNA Res. 2015, 22, 377–386. [Google Scholar] [CrossRef]
- Lecomte, L.; Arnyasi, M.; Ferchaud, A.L.; Kent, M.; Lien, S.; Stenlokk, K.; Sylvestre, F.; Bernatchez, L.; Merot, C. Investigating structural variant, indel and single nucleotide polymorphism differentiation between locally adapted Atlantic salmon populations. Evol. Appl. 2024, 17, e13653. [Google Scholar] [CrossRef]
- Vijayakumar, P.; Singaravadivelan, A.; Mishra, A.; Jagadeesan, K.; Bakyaraj, S.; Suresh, R.; Sivakumar, T. Whole-genome comparative analysis reveals genetic mechanisms of disease resistance and heat tolerance of tropical Bos indicus cattle breeds. Genome 2022, 65, 241–254. [Google Scholar] [CrossRef]
- Thambiraja, M.; Iyengar, S.K.; Satishkumar, B.; Kavuru, S.R.; Katari, A.; Singh, D.; Onteru, S.K.; Yennamalli, R.M. Genetic basis of immunity in Indian cattle as revealed by comparative analysis of Bos genome. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ben-Jemaa, S.; Boussaha, M.; Mandonnet, N.; Bardou, P.; Naves, M. Uncovering structural variants in Creole cattle from Guadeloupe and their impact on environmental adaptation through whole genome sequencing. PLoS ONE 2024, 19, e0309411. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, F.; Li, S.; Luo, X.; Peng, L.; Dong, Z.; Pausch, H.; Leonard, A.S.; Crysnanto, D.; Wang, S.; et al. Structural variation and introgression from wild populations in East Asian cattle genomes confer adaptation to local environment. Genome Biol. 2023, 24, 211. [Google Scholar] [CrossRef]
- Ayalew, W.; Wu, X.; Tarekegn, G.M.; Sisay Tessema, T.; Naboulsi, R.; Van Damme, R.; Bongcam-Rudloff, E.; Edea, Z.; Enquahone, S.; Yan, P. Whole-Genome Resequencing Reveals Selection Signatures of Abigar Cattle for Local Adaptation. Animals 2023, 13, 3269. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Stafuzza, N.B.; Machado, M.A.; do Carmo Panetto, J.C.; do Egito, A.A.; Baldi, F.; da Silva, M. Assessment of copy number variants in three Brazilian locally adapted cattle breeds using whole-genome re-sequencing data. Anim. Genet. 2023, 54, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.D.; Dzama, K.; Muchadeyi, F.C. Genetic Diversity of Seven Cattle Breeds Inferred Using Copy Number Variations. Front. Genet. 2018, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Niu, Y.; Chen, B.; Qin, P.; Wang, Y.; Hou, D.; Li, T.; Li, R.; Wang, C.; Yin, H.; et al. Genetic effect of an InDel in the promoter region of the NUDT15 and its effect on myoblast proliferation in chickens. BMC Genom. 2022, 23, 138. [Google Scholar] [CrossRef]
- Wheeler, M.M.; Stilp, A.M.; Rao, S.; Halldorsson, B.V.; Beyter, D.; Wen, J.; Mihkaylova, A.V.; McHugh, C.P.; Lane, J.; Jiang, M.Z.; et al. Whole genome sequencing identifies structural variants contributing to hematologic traits in the NHLBI TOPMed program. Nat. Commun. 2022, 13, 7592. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, C.; Ge, P.; Li, F.; Zhu, L.; Wang, Y.; Tao, J.; Zhang, X.; Dong, H.; Gai, W.; et al. A 21-bp InDel in the promoter of STP1 selected during tomato improvement accounts for soluble solid content in fruits. Hortic. Res. 2023, 10, uhad009. [Google Scholar] [CrossRef]
- Gebrie, A. Transposable elements as essential elements in the control of gene expression. Mob. DNA 2023, 14, 9. [Google Scholar] [CrossRef]
- Balachandran, P.; Walawalkar, I.A.; Flores, J.I.; Dayton, J.N.; Audano, P.A.; Beck, C.R. Transposable element-mediated rearrangements are prevalent in human genomes. Nat. Commun. 2022, 13, 7115. [Google Scholar] [CrossRef]
- Kelly, C.J.; Chitko-McKown, C.G.; Chuong, E.B. Ruminant-specific retrotransposons shape regulatory evolution of bovine immunity. Genome Res. 2022, 32, 1474–1486. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Han, X.; Han, J.; Hu, Y.; Li, F.; Xia, H.; Peng, L.; Boschiero, C.; Rosen, B.D.; et al. Assembly of a pangenome for global cattle reveals missing sequences and novel structural variations, providing new insights into their diversity and evolutionary history. Genome Res. 2022, 32, 1585–1601. [Google Scholar] [CrossRef]
- Shi, L.Y.; Zhang, P.; Yu, B.; Liu, Q.; Liu, C.H.; Lu, W.; Cheng, L.; Chen, H.B. Whole-Genome Sequencing Reveals the Role of Cis-Regulatory Elements and eQTL/sQTL in the Adaptive Selection of Hubei Indigenous Cattle. Animals 2025, 15, 1301. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhang, P.; Liu, Q.; Liu, C.; Cheng, L.; Yu, B.; Chen, H. Genome-Wide Analysis of Genetic Diversity and Selection Signatures in Zaobei Beef Cattle. Animals 2024, 14, 2447. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Yang, R.D.; Nelson, A.C.; Henzler, C.; Thyagarajan, B.; Silverstein, K.A.T. ScanIndel: A hybrid framework for indel detection via gapped alignment, split reads and assembly. Genome Med. 2015, 7, 127. [Google Scholar] [CrossRef]
- Pokrovac, I.; Pezer, Ä. Recent advances and current challenges in population genomics of structural variation in animals and plants. Front. Genet. 2022, 13, 1060898. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; van Binsbergen, R.; Brondum, R.F.; Liao, X.; Djari, A.; Rodriguez, S.C.; Grohs, C.; et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014, 46, 858–865. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Kallberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, Z.N.; Osborne, E.J.; Cone, K.R.; Kennedy, B.J.; Domyan, E.T.; Shapiro, M.D.; Elde, N.C.; Yandell, M. Wham: Identifying Structural Variants of Biological Consequence. PLoS Comput. Biol. 2015, 11, e1004572. [Google Scholar] [CrossRef] [PubMed]
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bahler, J.; Sedlazeck, F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017, 8, 14061. [Google Scholar] [CrossRef]
- Garrison, E.; Sirén, J.; Novak, A.M.; Hickey, G.; Eizenga, J.M.; Dawson, E.T.; Jones, W.; Garg, S.; Markello, C.; Lin, M.F.; et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 2018, 36, 875–879. [Google Scholar] [CrossRef]
- Sirén, J.; Monlong, J.; Chang, X.; Novak, A.M.; Eizenga, J.M.; Markello, C.; Sibbesen, J.A.; Hickey, G.; Chang, P.C.; Carroll, A.; et al. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science 2021, 374, abg8871. [Google Scholar] [CrossRef]
- Hickey, G.; Heller, D.; Monlong, J.; Sibbesen, J.A.; Sirén, J.; Eizenga, J.; Dawson, E.T.; Garrison, E.; Novak, A.M.; Paten, B. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 2020, 21, 35. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Y.; Pan, Y.; Xiang, X.; Peng, C.; He, J.; Huang, G.; Wang, Z.; Zhao, P. Genomic insights into demographic history, structural variation landscape, and complex traits from 514 Hu sheep genomes. J. Genet. Genom. 2025, 52, 245–257. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef] [PubMed]
- Kern, C.; Wang, Y.; Xu, X.; Pan, Z.; Halstead, M.; Chanthavixay, G.; Saelao, P.; Waters, S.; Xiang, R.; Chamberlain, A.; et al. Functional annotations of three domestic animal genomes provide vital resources for comparative and agricultural research. Nat. Commun. 2021, 12, 1821. [Google Scholar] [CrossRef] [PubMed]
- Gel, B.; Díez-Villanueva, A.; Serra, E.; Buschbeck, M.; Peinado, M.A.; Malinverni, R. regioneR: An R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 2016, 32, 289–291. [Google Scholar] [CrossRef]
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.J.; Pairo-Castineira, E.; et al. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef]
- Elizarraras, J.M.; Liao, Y.; Shi, Z.; Zhu, Q.; Pico, A.R.; Zhang, B. WebGestalt 2024: Faster gene set analysis and new support for metabolomics and multi-omics. Nucleic Acids Res. 2024, 52, W415–W421. [Google Scholar] [CrossRef]
- Talenti, A.; Powell, J.; Wragg, D.; Chepkwony, M.; Fisch, A.; Ferreira, B.R.; Mercadante, M.E.Z.; Santos, I.M.; Ezeasor, C.K.; Obishakin, E.T.; et al. Optical mapping compendium of structural variants across global cattle breeds. Sci. Data 2022, 9, 618. [Google Scholar] [CrossRef]
- Guan, X.W.; Zhao, S.P.; Xiang, W.X.; Jin, H.; Chen, N.B.; Lei, C.Z.; Jia, Y.T.; Xu, L. Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing. Biology 2022, 11, 1327. [Google Scholar] [CrossRef]
- de la Chaux, N.; Messer, P.W.; Arndt, P.F. DNA indels in coding regions reveal selective constraints on protein evolution in the human lineage. Bmc Evol. Biol. 2007, 7, 191. [Google Scholar] [CrossRef]
- Yang, Y.; Braga, M.; Dean, M.D. Insertion-Deletion Events Are Depleted in Protein Regions with Predicted Secondary Structure. Genome Biol. Evol. 2024, 16, evae093. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.H.; Chen, L.; Xiong, Y.L.; Chen, Z.X. Evolution and function of developmentally dynamic pseudogenes in mammals. Genome Biol. 2022, 23, 235. [Google Scholar] [CrossRef] [PubMed]
- Tutar, Y. Pseudogenes. Comp. Funct. Genom. 2012, 2012, 424526. [Google Scholar] [CrossRef]
- Esnault, C.; Maestre, J.; Heidmann, T. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 2000, 24, 363–367. [Google Scholar] [CrossRef]
- Zhang, Z.; Carriero, N.; Gerstein, M. Comparative analysis of processed pseudogenes in the mouse and human genomes. Trends Genet. 2004, 20, 62–67. [Google Scholar] [CrossRef]
- Cameron, R.A.; Chow, S.H.; Berney, K.; Chiu, T.Y.; Yuan, Q.A.; Kramer, A.; Helguero, A.; Ransick, A.; Yun, M.; Davidson, E.H. An evolutionary constraint: Strongly disfavored class of change in DNA sequence during divergence of cis-regulatory modules. Proc. Natl. Acad. Sci. USA 2005, 102, 11769–11774. [Google Scholar] [CrossRef]
- Martinez, C.; Rest, J.S.; Kim, A.R.; Ludwig, M.; Kreitman, M.; White, K.; Reinitz, J. Ancestral resurrection of the Drosophila S2E enhancer reveals accessible evolutionary paths through compensatory change. Mol. Biol. Evol. 2014, 31, 903–916. [Google Scholar] [CrossRef]
- Barriere, A.; Gordon, K.L.; Ruvinsky, I. Coevolution within and between regulatory loci can preserve promoter function despite evolutionary rate acceleration. PLoS Genet. 2012, 8, e1002961. [Google Scholar] [CrossRef]
- Kliesmete, Z.; Orchard, P.; Lee, V.Y.K.; Geuder, J.; Krauss, S.M.; Ohnuki, M.; Jocher, J.; Vieth, B.; Enard, W.; Hellmann, I. Evidence for compensatory evolution within pleiotropic regulatory elements. Genome Res. 2024, 34, 1528–1539. [Google Scholar] [CrossRef]
- Chiang, C.; Scott, A.J.; Davis, J.R.; Tsang, E.K.; Li, X.; Kim, Y.; Hadzic, T.; Damani, F.N.; Ganel, L.; Montgomery, S.B.; et al. The impact of structural variation on human gene expression. Nat. Genet. 2017, 49, 692–699. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.Y.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Ruderfer, D.M.; Hamamsy, T.; Lek, M.; Karczewski, K.J.; Kavanagh, D.; Samocha, K.E.; Daly, M.J.; MacArthur, D.G.; Fromer, M.; Purcell, S.M.; et al. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat. Genet. 2016, 48, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef]
- Gemayel, R.; Cho, J.; Boeynaems, S.; Verstrepen, K.J. Beyond Junk-Variable Tandem Repeats as Facilitators of Rapid Evolution of Regulatory and Coding Sequences. Genes 2012, 3, 461–480. [Google Scholar] [CrossRef]
- Miller, C.J.; Usdin, K. Mismatch repair is a double-edged sword in the battle against microsatellite instability. Expert. Rev. Mol. Med. 2022, 24, e32. [Google Scholar] [CrossRef]
- Lawson, H.A.; Liang, Y.H.; Wang, T. Transposable elements in mammalian chromatin organization. Nat. Rev. Genet. 2023, 24, 712–723. [Google Scholar] [CrossRef]
- Qiu, X.; Qin, X.; Chen, L.; Chen, Z.; Hao, R.; Zhang, S.; Yang, S.; Wang, L.; Cui, Y.; Li, Y.; et al. Serum Biochemical Parameters, Rumen Fermentation, and Rumen Bacterial Communities Are Partly Driven by the Breed and Sex of Cattle When Fed High-Grain Diet. Microorganisms 2022, 10, 323. [Google Scholar] [CrossRef]
- Wei, M.; Liu, X.; Xie, P.; Lei, Y.; Yu, H.; Han, A.; Xie, L.; Jia, H.; Lin, S.; Bai, Y.; et al. Characterization of Volatile Profiles and Correlated Contributing Compounds in Pan-Fried Steaks from Different Chinese Yellow Cattle Breeds through GC-Q-Orbitrap, E-Nose, and Sensory Evaluation. Molecules 2022, 27, 3593. [Google Scholar] [CrossRef]
- Luo, C.; Xu, X.; Zhao, C.; Wang, Q.; Wang, R.; Lang, D.; Zhang, J.; Hu, W.; Mu, Y. Insight Into Body Size Evolution in Aves: Based on Some Body Size-Related Genes. Integr. Zool. 2024; in press. [Google Scholar] [CrossRef]
- Deng, M.T.; Zhu, F.; Yang, Y.Z.; Yang, F.X.; Hao, J.P.; Chen, S.R.; Hou, Z.C. Genome-wide association study reveals novel loci associated with body size and carcass yields in Pekin ducks. BMC Genom. 2019, 20, 1. [Google Scholar] [CrossRef]
- Janssens, B.; Mohapatra, B.; Vatta, M.; Goossens, S.; Vanpoucke, G.; Kools, P.; Montoye, T.; van Hengel, J.; Bowles, N.E.; van Roy, F.; et al. Assessment of the CTNNA3 gene encoding human alpha T-catenin regarding its involvement in dilated cardiomyopathy. Hum. Genet. 2003, 112, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, F.; Yuan, L.; Zhang, X.; Zhang, D.; Li, X.; Zhang, Y.; Zhao, Y.; Song, Q.; Wang, J.; et al. Expression of ovine CTNNA3 and CAP2 genes and their association with growth traits. Gene 2022, 807, 145949. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Niu, Q.; Jiang, J.; Wang, G.; Zhou, P.; Li, J.; Chen, C.; Liu, L.; Xu, L.; Ren, H. Identifying Candidate Genes for Litter Size and Three Morphological Traits in Youzhou Dark Goats Based on Genome-Wide SNP Markers. Genes 2023, 14, 1183. [Google Scholar] [CrossRef]
- Yu, H.W.; Yu, S.C.; Guo, J.T.; Cheng, G.; Mei, C.G.; Zan, L.S. Genome-Wide Association Study Reveals Novel Loci Associated with Body Conformation Traits in Qinchuan Cattle. Animals 2023, 13, 3628. [Google Scholar] [CrossRef]
- Del Gobbo, G.F.; Yin, Y.; Choufani, S.; Butcher, E.A.; Wei, J.; Rajcan-Separovic, E.; Bos, H.; von Dadelszen, P.; Weksberg, R.; Robinson, W.P.; et al. Genomic imbalances in the placenta are associated with poor fetal growth. Mol. Med. 2021, 27, 3. [Google Scholar] [CrossRef]
- Teodoro, M.; Maiorano, A.M.; Campos, G.S.; de Albuquerque, L.G.; de Oliveira, H.N. Genetic parameters, genomic prediction, and identification of regulatory regions located on chromosome 14 for weight traits in Nellore cattle. J. Anim. Breed. Genet. 2025, 142, 184–199. [Google Scholar] [CrossRef]
- Alam, M.Z.; Haque, M.A.; Iqbal, A.; Lee, Y.M.; Ha, J.J.; Jin, S.; Park, B.; Kim, N.Y.; Won, J.I.; Kim, J.J. Genome-Wide Association Study to Identify QTL for Carcass Traits in Korean Hanwoo Cattle. Animals 2023, 13, 2737. [Google Scholar] [CrossRef]
- Haque, M.A.; Lee, Y.M.; Ha, J.J.; Jin, S.; Park, B.; Kim, N.Y.; Won, J.I.; Kim, J.J. Genome-wide association study identifies genomic regions associated with key reproductive traits in Korean Hanwoo cows. BMC Genom. 2024, 25, 496. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, J.; Wang, J.; Zhang, L.; Xu, L.; Chen, Y.; Zhu, B.; Wang, Z.; Gao, H.; Li, J.; et al. Genome-Wide Detection of Copy Number Variations and Their Potential Association with Carcass and Meat Quality Traits in Pingliang Red Cattle. Int. J. Mol. Sci. 2024, 25, 5626. [Google Scholar] [CrossRef]
- Jahromi, M.M. Haplotype specific alteration of diabetes MHC risk by olfactory receptor gene polymorphism. Autoimmun. Rev. 2012, 12, 270–274. [Google Scholar] [CrossRef]
- Tan, Y.; Yang, S.; Liu, Q.; Li, Z.; Mu, R.; Qiao, J.; Cui, L. Pregnancy-related complications in systemic lupus erythematosus. J. Autoimmun. 2022, 132, 102864. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.; Davison, J.M. Type 1 diabetes and pregnancy. BMJ 2007, 334, 742–745. [Google Scholar] [CrossRef]
- Wu, Q.d.; Zhou, Y.d.; Wang, Y.; Zhang, Y.; Shen, Y.; Su, Q.; Gao, G.; Xu, H.; Zhou, X.; Liu, B. Whole-genome sequencing reveals breed-differential CNVs between Tongcheng and Large White pigs. Anim. Genet. 2020, 51, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Notay, K.; Downey, G.P.; McCulloch, C.A. The Leucine-Rich Repeat Region of CARMIL1 Regulates IL-1-Mediated ERK Activation, MMP Expression, and Collagen Degradation. Cell Rep. 2020, 31, 107781. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Qu, K.; Ma, Z.; Zhan, J.; Zhang, F.; Shen, J.; Ning, Q.; Jia, P.; Zhang, J.; Chen, N.; et al. Genome-Wide Association Study Identifies Genomic Loci Associated With Neurotransmitter Concentration in Cattle. Front. Genet. 2020, 11, 139. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison Breed | Shared Genes |
|---|---|
| Wuling vs. Dabieshan | RUNX1, TRPM3, SHISAL2A |
| Wuling vs. Yunba | UBXN2B, GLRA3 |
| Wuling vs. Yiling | TLN2, UBXN2B |
| Wuling vs. Zaobei | AKAP10, RUNX1, LRRC7, LAMA2, PIGL, PLD1, USP25, ANO3, PLD5, MTHFD2L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, L.; Zhang, P.; Yu, B.; Cheng, L.; Liu, S.; Liu, Q.; Zhou, Y.; Xiang, M.; Zhao, P.; Chen, H. Genomic Analysis of Indel and SV Reveals Functional and Adaptive Signatures in Hubei Indigenous Cattle Breeds. Animals 2025, 15, 1755. https://doi.org/10.3390/ani15121755
Shi L, Zhang P, Yu B, Cheng L, Liu S, Liu Q, Zhou Y, Xiang M, Zhao P, Chen H. Genomic Analysis of Indel and SV Reveals Functional and Adaptive Signatures in Hubei Indigenous Cattle Breeds. Animals. 2025; 15(12):1755. https://doi.org/10.3390/ani15121755
Chicago/Turabian StyleShi, Liangyu, Pu Zhang, Bo Yu, Lei Cheng, Sha Liu, Qing Liu, Yuan Zhou, Min Xiang, Pengju Zhao, and Hongbo Chen. 2025. "Genomic Analysis of Indel and SV Reveals Functional and Adaptive Signatures in Hubei Indigenous Cattle Breeds" Animals 15, no. 12: 1755. https://doi.org/10.3390/ani15121755
APA StyleShi, L., Zhang, P., Yu, B., Cheng, L., Liu, S., Liu, Q., Zhou, Y., Xiang, M., Zhao, P., & Chen, H. (2025). Genomic Analysis of Indel and SV Reveals Functional and Adaptive Signatures in Hubei Indigenous Cattle Breeds. Animals, 15(12), 1755. https://doi.org/10.3390/ani15121755