Variations in Genetic Diversity of Invasive Species Lithobates catesbeianus in China


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
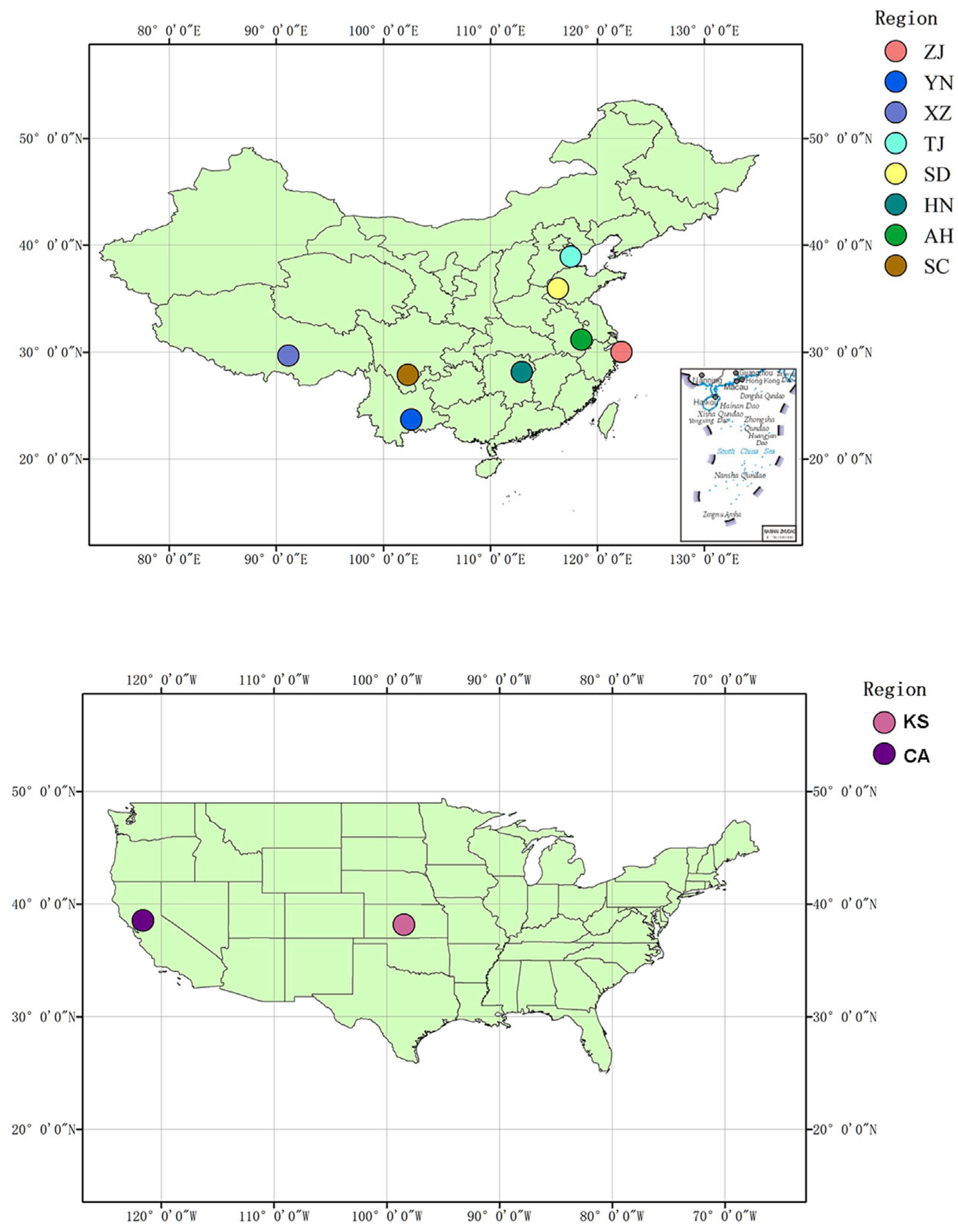
2.2. Study Area and Sampling for Bullfrogs and DNA Extractions
2.3. Microsatellite Genotyping
2.4. Genome Sequencing, Alignment, and SNP Calling
2.5. Data Analyses
2.5.1. Microsatellites
2.5.2. SNP Data
3. Results
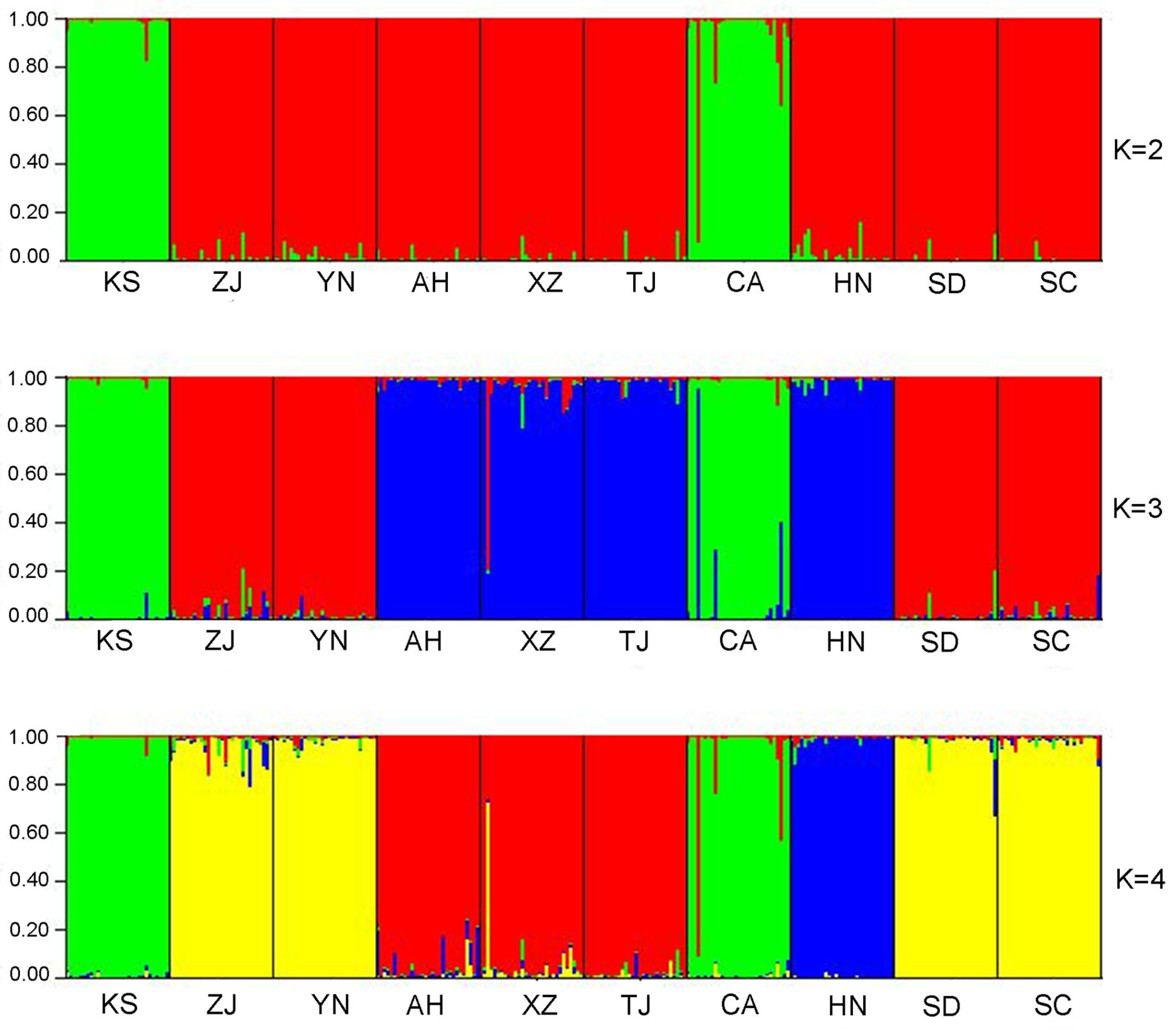
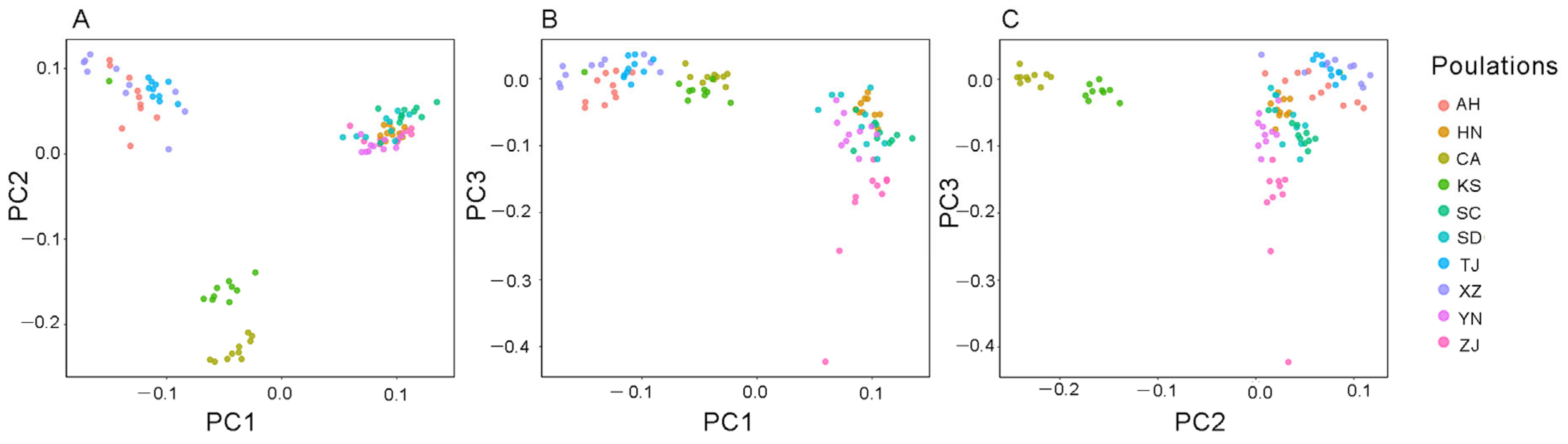
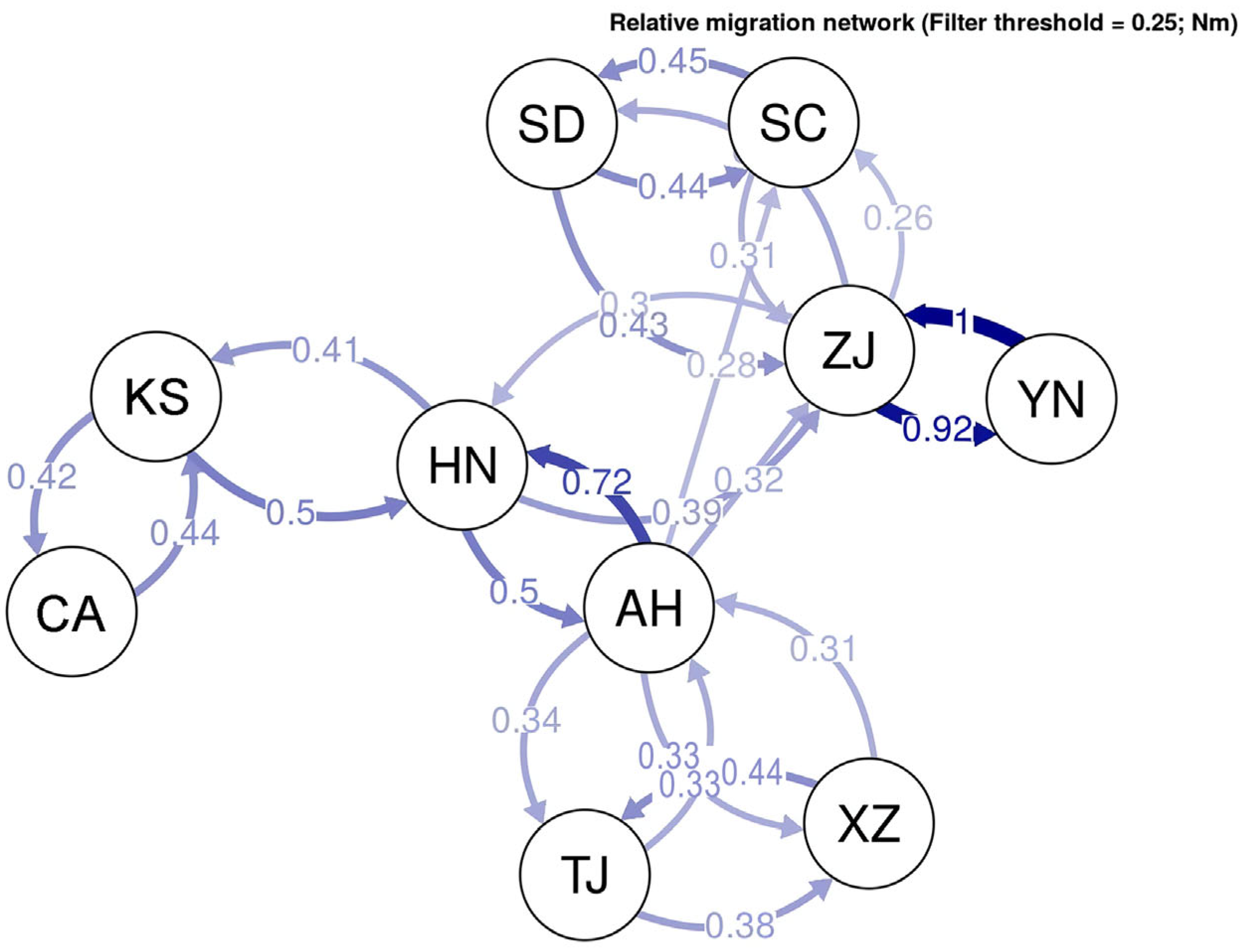
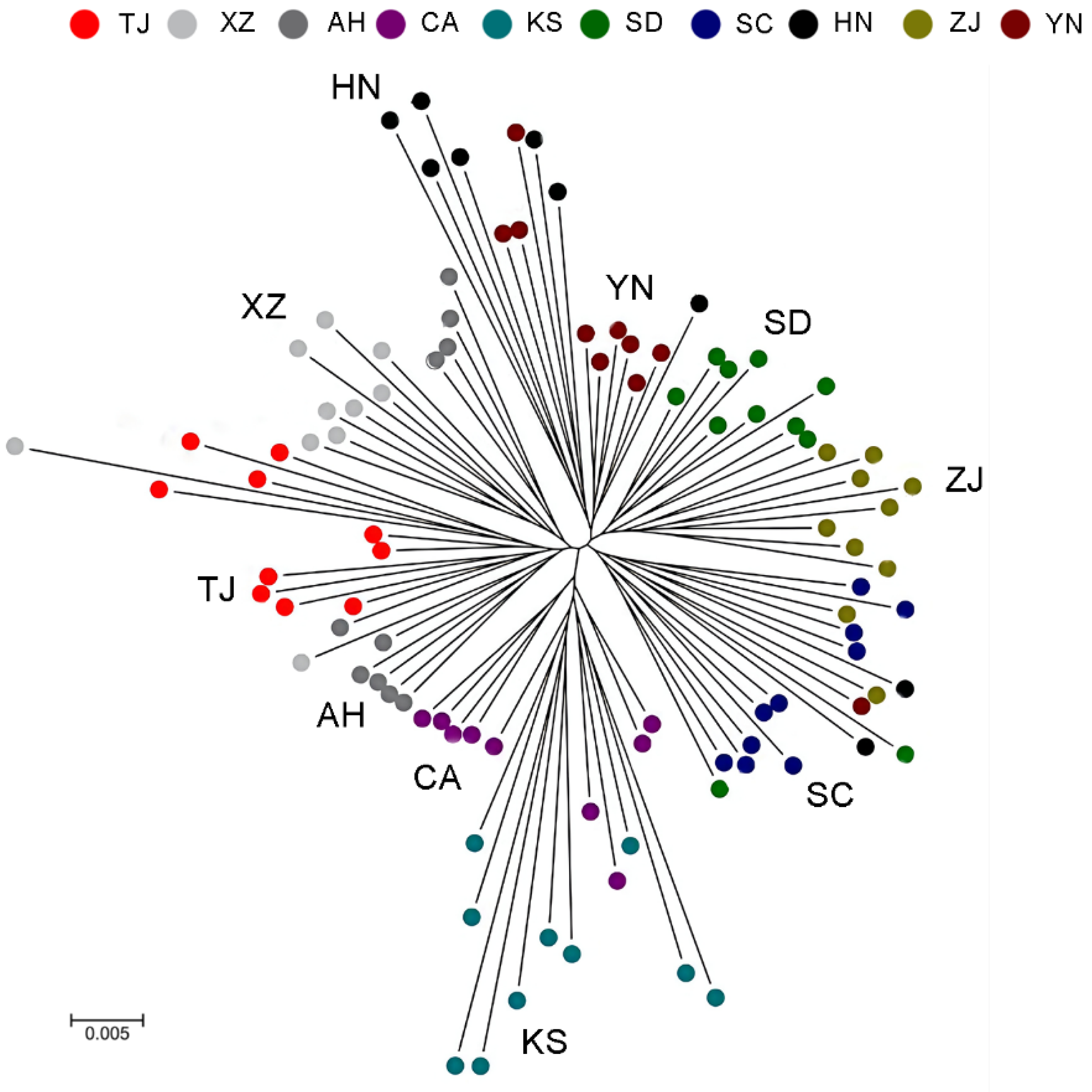
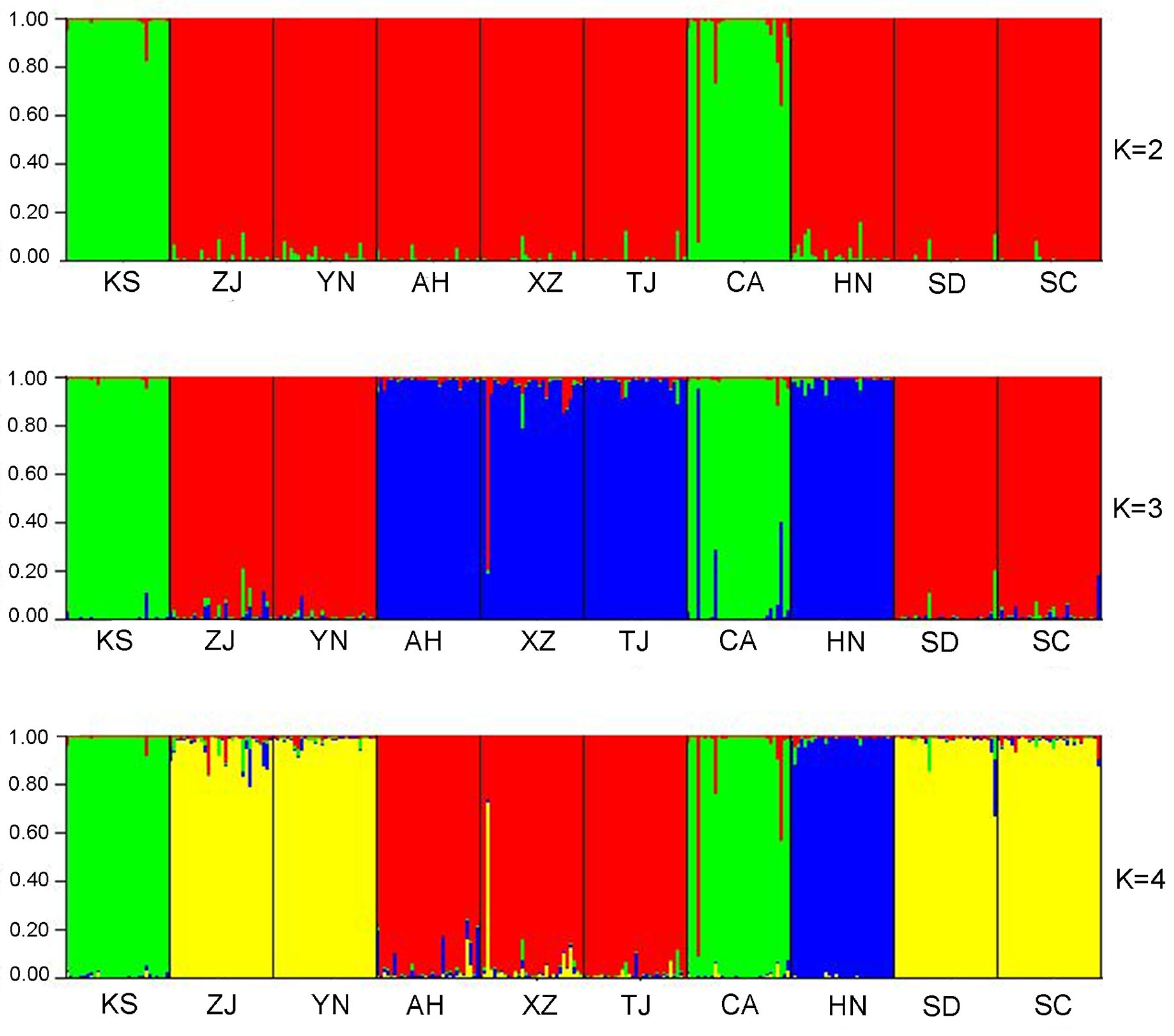
3.1. Microsatellite Genetic Structure in Chinese Bullfrog Populations
3.2. Genome Sequencing and SNP Calling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pimentel, D.; Lach, L.; Zuniga, R.; Morrison, D. Environmental and Economic Costs of Nonindigenous Species in the United States. BioScience 2000, 50, 53–65. [Google Scholar] [CrossRef]
- Pimentel, D.; Zuniga, R.; Morrison, D. Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecol. Econ. 2005, 52, 273–288. [Google Scholar] [CrossRef]
- Bellard, C.; Genovesi, P.; Jeschke, J.M. Global patterns in threats to vertebrates by biological invasions. Proc. Biol. Sci. 2016, 283, 20152454. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Brunet, J.; Guries, R.P. The extent of hybridization and its impact on the genetic diversity and population structure of an invasive tree, Ulmus pumila (Ulmaceae). Evol. Appl. 2010, 3, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.E.; Smith, G.C.; Jones, S.M. Is anti-predator behaviour in Tasmanian eastern quolls (Dasyurus viverrinus) effective against introduced predators? Anim. Conserv. 2004, 7, 155–160. [Google Scholar] [CrossRef]
- Kiesecker, J.M.; Blaustein, A.R.; Miller, C.L. Potential Mechanisms Underlying the Displacement of Native Red-Legged Frogs by Introduced Bullfrogs. Ecology 2001, 82, 1964–1970. [Google Scholar] [CrossRef]
- Mack, R.N.; Simberloff, D.; Lonsdale, W.M.; Evans, H.; Clout, M.; Bazzaz, F.A. Biotic invasions causes, epidemiology, global consequences, and control. Issues Ecol. 2000, 10, 689–710. [Google Scholar] [CrossRef]
- Juliano, S.A.; Philip Lounibos, L. Ecology of invasive mosquitoes: Effects on resident species and on human health. Ecol. Lett. 2005, 8, 558–574. [Google Scholar] [CrossRef]
- Thompson, M.E.; Nowakowski, A.J.; Donnelly, M.A. The importance of defining focal assemblages when evaluating amphibian and reptile responses to land use. Conserv. Biol. 2016, 30, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, I.; Andreychev, A.; Kulakhmetov, R.; Lobachev, E. Commensals of underground mammals: European mole (Talpa europaea, Eulipotyphla, Talpidae) and the greater mole-rat (Spalax microphthalmus, Rodentia, Spalacidae). Biodiversitas J. Biol. Divers. 2021, 22, 4665–4670. [Google Scholar] [CrossRef]
- Xian, X.; Chen, H.; Zhao, J. Introduction to China Invasive Alien Species Database. 2013, Volume 39. Available online: www.chinaias.cn (accessed on 20 June 2023).
- Wang, K.; Ren, J.; Chen, H.; Lyu, Z.; Guo, X.; Jiang, K.; Chen, J.; Li, J.; Guo, P.; Wang, Y.; et al. The updated checklists of amphibians and reptiles of China. Biodivers. Sci. 2020, 28, 189. [Google Scholar]
- ISSG. Invasive Species Specialist Group. 2008. Available online: http://www.issg.org/database/welcome/ (accessed on 6 June 2011).
- Liu, X.; Wang, S.; Ke, Z.; Cheng, C.; Wang, Y.; Zhang, F.; Xu, F.; Li, X.; Gao, X.; Jin, C.; et al. More invaders do not result in heavier impacts: The effects of non- native bullfrogs on native anurans are mitigated by high densities of non- native crayfish. J. Anim. Ecol. 2018, 87, 850–862. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.P.; Dritz, D.; Strange, T.; Holyoak, M. Effects of introduced mosquitofish and bullfrogs on the threatened California red-legged frog. Conserv. Biol. 1999, 13, 613–622. [Google Scholar] [CrossRef]
- Garner, T.W.; Perkins, M.W.; Govindarajulu, P.; Seglie, D.; Walker, S.; Cunningham, A.A.; Fisher, M.C. The emerging amphibian pathogen Batrachochytrium dendrobatidis globally infects introduced populations of the North American bullfrog, Rana catesbeiana. Biol. Lett. 2006, 2, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Boone, M.D.; Little, E.E.; Semlitsch, R.D.; Fox, S.F. Overwintered Bullfrog Tadpoles Negatively Affect Salamanders and Anurans in Native Amphibian Communities. Copeia 2004, 2004, 683–690. [Google Scholar] [CrossRef]
- Pearl, C.A.; Adams, M.J.; Leuthold, N.; Bury, R.B. Amphibian occurrence and aquatic invaders in a changing landscape: Implications for wetland mitigation in the Willamette Valley, Oregon, USA. Wetlands 2005, 25, 76–88. [Google Scholar] [CrossRef]
- Kraus, F. Alien Reptiles and Amphibians: A Scientific Compendium and Analysis; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Ficetola, G.F.; Thuiller, W.; Miaud, C. Prediction and validation of the potential global distribution of a problematic alien invasive species—The American bullfrog. Divers. Distrib. 2007, 13, 476–485. [Google Scholar] [CrossRef]
- Liu, X.; McGarrity, M.E.; Bai, C.; Ke, Z.; Li, Y. Ecological knowledge reduces religious release of invasive species. Ecosphere 2013, 4, art21. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, F.; Xu, F.; Li, X.; Gao, X.; Jin, C.; Zhu, W.; Yan, S.; Li, Y. Habitat Selection by the Introduced American Bullfrog (Lithobates catesbeianus) on Daishan Island, China. BioOne 2009, 43, 205–211. [Google Scholar] [CrossRef]
- Zeng, X. Prospect and strategy of bullfrog farming. Inland Fish 1998, 10, 4–5. [Google Scholar]
- Liu, X.; Li, Y. Aquaculture enclosures relate to the establishment of feral populations of introduced species. PLoS ONE 2009, 4, e6199. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Ke, Z.; Consuegra, S.; Liu, X.; Li, Y. The role of founder effects on the genetic structure of the invasive bullfrog (Lithobates catesbeianaus) in China. Biol. Invasions 2012, 14, 1785–1796. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Xu, C.; Wang, S.; Du, J.; Niu, M.; Yang, J.; Li, Y. Pathogenic selection promotes adaptive immune variations against serious bottlenecks in early invasions of bullfrogs. Iscience 2023, 26, 107316. [Google Scholar] [CrossRef] [PubMed]
- Hamelin, R.C.; Roe, A.D. Genomic biosurveillance of forest invasive alien enemies: A story written in code. Evol. Appl. 2020, 13, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Resh, C.A.; Galaska, M.P.; Benesh, K.C.; Gardner, J.P.A.; Wei, K.-J.; Yan, R.-J.; Mahon, A.R. Using Genomics to Link Populations of an Invasive Species to Its Potential Sources. Front. Ecol. Evol. 2021, 9, 575599. [Google Scholar] [CrossRef]
- Wang, S.; Liu, C.; Wu, J.; Xu, C.; Zhang, J.; Bai, C.; Gao, X.; Li, X.; Li, X.; Zhu, W.; et al. Propagule pressure and hunting pressure jointly determine genetic evolution in insular populations of a global frog invader. Sci. Rep. 2019, 9, 448. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.D.; Davila, J.A.; Lougheed, S.C.; Boag, P.T. Genetic evidence for female-biased dispersal in the bullfrog, Rana catesbeiana (Ranidae). Mol. Ecol. 2003, 12, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Wang, S.; Meyer, E.; McKay, J.K.; Matz, M.V. 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat. Methods 2012, 9, 808–810. [Google Scholar] [CrossRef]
- Hammond, S.A.; Warren, R.L.; Vandervalk, B.P.; Kucuk, E.; Khan, H.; Gibb, E.A.; Pandoh, P.; Kirk, H.; Zhao, Y.; Jones, M.; et al. The North American bullfrog draft genome provides insight into hormonal regulation of long noncoding RNA. Nat. Commun. 2017, 8, 1433. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. Population Genetics Software for Exact Tests and Ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rice, W. Analyzing table of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.O.D.; Smouse, P.E. genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Beerli, P.; Felsenstein, J. Maximum-likelihood estimation of migration rates and effective population numbers in two. Genetics 1999, 152, 763. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Ravigne, V.; Estoup, A. Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1.0). BMC Bioinform. 2010, 11, 401. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Yang, J. Kernel feature extraction methods observed from the viewpoint of generating-kernels. Front. Electr. Electron. Eng. China 2011, 6, 43–55. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BioMed Cent. 2011, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A.; O’Hara, R.B. diveRsity: AnRpackage for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.M.; Trexler, J.C.; Nico, L.G.; Rawlings, T.A. Genetic Diversity in a Morphologically Conservative Invasive Taxon: Multiple Introductions of Swamp Eels to the Southeastern United States. Conserv. Biol. 2002, 16, 1024–1035. [Google Scholar] [CrossRef]
- Hierro, J.L.; Maron, J.L.; Callaway, R.M. A biogeographical approach to plant invasions: The importance of studying exotics in their introduced and native range. J. Ecol. 2004, 93, 5–15. [Google Scholar] [CrossRef]
- Bock, D.G.; Caseys, C.; Cousens, R.D.; Hahn, M.A.; Heredia, S.M.; Hubner, S.; Turner, K.G.; Whitney, K.D.; Rieseberg, L.H. What we still don’t know about invasion genetics. Mol. Ecol. 2015, 24, 2277–2297. [Google Scholar] [CrossRef] [PubMed]
- Wellband, K.W.; Pettitt-Wade, H.; Fisk, A.T.; Heath, D.D. Standing genetic diversity and selection at functional gene loci are associated with differential invasion success in two non-native fish species. Mol. Ecol. 2018, 27, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, N.D.; Suarez, A.V.; Holway, D.A.; Case, T.J. Reduced genetic variation and the success of an invasive species. Proc. Natl. Acad. Sci. USA 2000, 97, 5948–5953. [Google Scholar] [CrossRef] [PubMed]
- Ficetola, G.F.; Bonin, A.; Miaud, C. Population genetics reveals origin and number of founders in a biological invasion. Mol. Ecol. 2008, 17, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Ficetola, G.F.; Garner, T.W.; Wang, J.; De Bernardi, F. Rapid selection against inbreeding in a wild population of a rare frog. Evol. Appl. 2011, 4, 30–38. [Google Scholar] [CrossRef] [PubMed]
- de Longevialle, A.F.; Meyer, E.H.; Andres, C.; Taylor, N.L.; Lurin, C.; Millar, A.H.; Small, I.D. The pentatricopeptide repeat gene OTP43 is required for trans-splicing of the mitochondrial nad1 Intron 1 in Arabidopsis thaliana. Plant Cell 2007, 19, 3256–3265. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.L.; Ge, S. Molecular phylogeny of Oryzeae (Poaceae) based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes. Am. J. Bot. 2005, 92, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Thang, T.N.; Vázquez-Prieto, S.; Vilas, R.; Paniagua, E.; Ubeira, F.M.; Ichikawa-Seki, M. Genetic diversity of Fasciola hepatica in Spain and Peru. Parasitol. Int. 2020, 76, 102100. [Google Scholar] [CrossRef] [PubMed]
- Irisarri, I.; Mauro, D.S.; Abascal, F.; Ohler, A.; Vences, M.; Zardoya, R. The origin of modern frogs (Neobatrachia) was accompanied by acceleration in mitochondrial and nuclear substitution rates. BMC Genom. 2012, 13, 626. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Butler, J.; Patterson, N.; Capelli, C.; Pascali, V.L.; Scarnicci, F.; Ruiz-Linares, A.; Groop, L.; Saetta, A.A.; Korkolopoulou, P. Discerning the ancestry of European Americans in genetic association studies. PLoS Genet. 2008, 4, e236. [Google Scholar] [CrossRef] [PubMed]
- Dlugosch, K.M.; Parker, I.M. Invading populations of an ornamental shrub show rapid life history evolution despite genetic bottlenecks. Ecol. Lett. 2008, 11, 701–709. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP Data | Microsatellite Data | |||||
|---|---|---|---|---|---|---|
| Location | Private Alleles | Ho | He | Na | Ho | He |
| Kansas | 17,304 | 0.13 | 0.12 | 6.7 ± 0.87 | 0.62 ± 0.07 | 0.6 ± 0.04 |
| California | 15,581 | 0.12 | 0.14 | 6.411 ± 1 | 0.62 ± 0.07 | 0.57 ± 0.06 |
| Hunan | 9207 | 0.10 | 0.12 | 5.47 ± 0.63 | 0.68 ± 0.07 | 0.57 ± 0.06 |
| Yunnan | 5239 | 0.06 | 0.11 | 5.23 ± 0.55 | 0.7 ± 0.05 | 0.61 ± 0.05 |
| Anhui | 6767 | 0.07 | 0.10 | 5.05 ± 0.67 | 0.79 ± 0.06 | 0.6 ± 0.04 |
| Tibet | 5117 | 0.09 | 0.09 | 4.88 ± 0.54 | 0.66 ± 0.08 | 0.53 ± 0.06 |
| Tianjin | 4330 | 0.08 | 0.08 | 4.76 ± 0.48 | 0.66 ± 0.06 | 0.58 ± 0.04 |
| Zhejiang | 5382 | 0.10 | 0.11 | 4.47 ± 0.35 | 0.68 ± 0.06 | 0.6 ± 0.04 |
| Shandong | 7408 | 0.10 | 0.09 | 4.35 ± 0.49 | 0.65 ± 0.09 | 0.52 ± 0.06 |
| Sichuan | 9809 | 0.08 | 0.10 | 4.12 ± 0.48 | 0.73 ± 0.06 | 0.58 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Xu, C.; Wang, S.; Wang, S.; Li, Y. Variations in Genetic Diversity of Invasive Species Lithobates catesbeianus in China. Animals 2024, 14, 1287. https://doi.org/10.3390/ani14091287
Zhang J, Xu C, Wang S, Wang S, Li Y. Variations in Genetic Diversity of Invasive Species Lithobates catesbeianus in China. Animals. 2024; 14(9):1287. https://doi.org/10.3390/ani14091287
Chicago/Turabian StyleZhang, Jiaqi, Chunxia Xu, Supen Wang, Siqi Wang, and Yiming Li. 2024. "Variations in Genetic Diversity of Invasive Species Lithobates catesbeianus in China" Animals 14, no. 9: 1287. https://doi.org/10.3390/ani14091287
APA StyleZhang, J., Xu, C., Wang, S., Wang, S., & Li, Y. (2024). Variations in Genetic Diversity of Invasive Species Lithobates catesbeianus in China. Animals, 14(9), 1287. https://doi.org/10.3390/ani14091287