
Genomic Inbreeding and Runs of Homozygosity Analysis of Cashmere Goat
 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Population, DNA Extraction and Whole-Genome Resequencing
2.2. The Processing of Whole-Genome Sequencing Data
2.3. The Analysis of Population Structure and Linkage Disequilibrium
2.4. Identification of ROH
2.5. ROH Classification and Assessment of Inbreeding Coefficients
2.6. Identification of Genes within ROH Islands of Domestic Goat Populations
2.7. Identification of Consensus Selection Signature Regions in Cashmere Goat Populations
3. Results
3.1. Population Structure Analyses and Linkage Disequilibrium
3.2. Distribution of Runs of Homozygosity
3.3. Inbreeding Coefficients
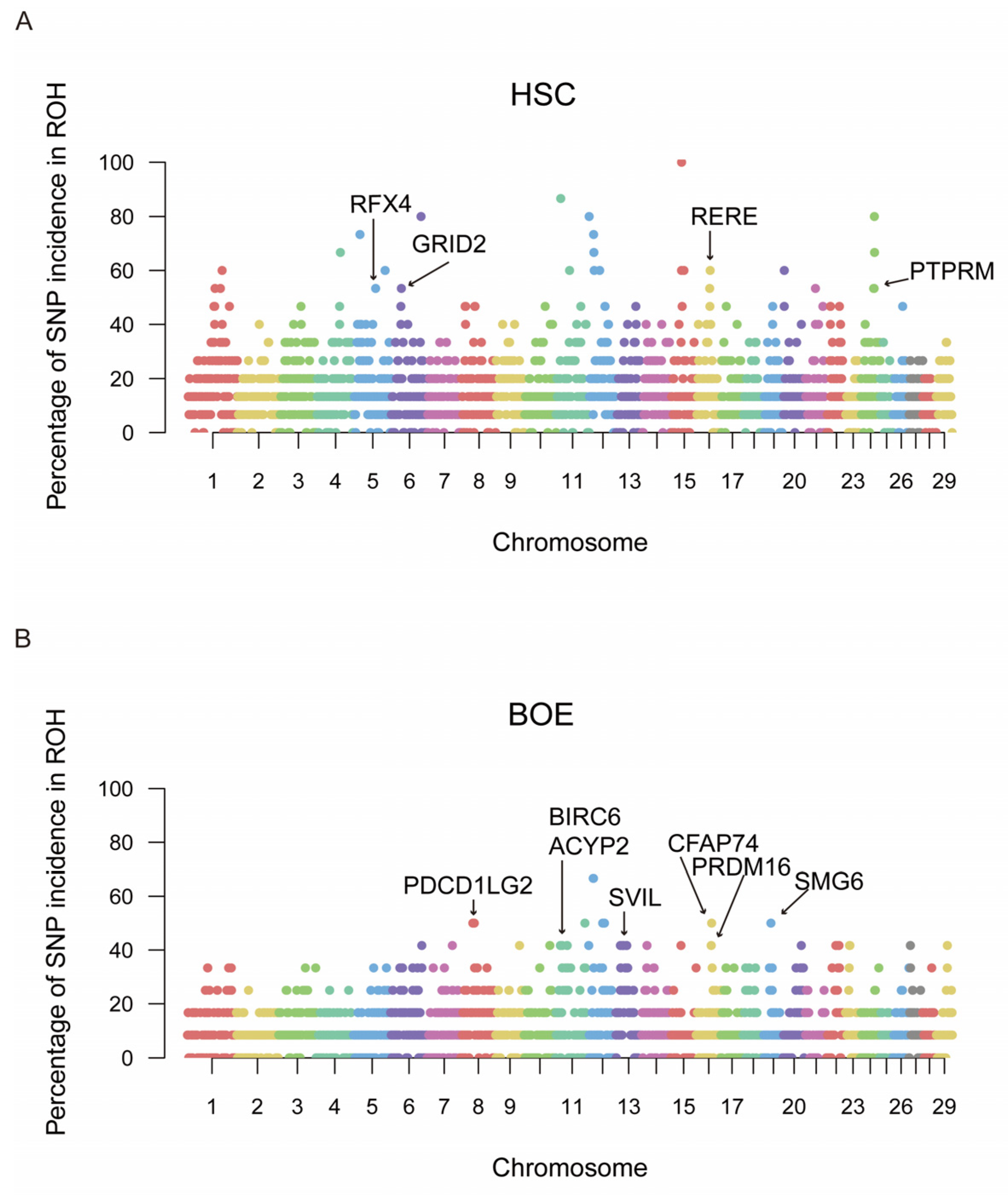
3.4. ROH Islands of Domestic Goat Populations
3.5. The Consensus ROH of IMC and HSC Breeds
4. Discussion
4.1. Population Structure and Linkage Disequilibrium
4.2. Patterns of Runs of Homozygosity (ROHs)
4.3. Inbreeding Levels within Populations
4.4. Functional Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Sumreddee, P.; Hay, E.H.; Toghiani, S.; Roberts, A.; Aggrey, S.E.; Rekaya, R. Grid search approach to discriminate between old and recent inbreeding using phenotypic, pedigree and genomic information. BMC Genom. 2021, 22, 538. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wang, L.; Liu, J.; Deng, T.; Yan, H.; Zhang, L.; Liu, X.; Gao, H.; Hou, X.; Wang, L.; et al. Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a Large White pig population. J. Anim. Sci. Biotechnol. 2020, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Metzger, J.; de Lemos, M.V.A.; Stafuzza, N.B.; Kluska, S.; Olivieri, B.F.; Feitosa, F.L.B.; Berton, M.P.; Lopes, F.B.; Munari, D.P.; et al. Autozygosity islands and ROH patterns in Nellore lineages: Evidence of selection for functionally important traits. BMC Genom. 2018, 19, 680. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.W.; Weber, J.L. Long homozygous chromosomal segments in reference families from the centre d’Etude du polymorphisme humain. Am. J. Hum. Genet. 1999, 65, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef]
- Nishio, M.; Inoue, K.; Ogawa, S.; Ichinoseki, K.; Arakawa, A.; Fukuzawa, Y.; Okamura, T.; Kobayashi, E.; Taniguchi, M.; Oe, M.; et al. Comparing pedigree and genomic inbreeding coefficients, and inbreeding depression of reproductive traits in Japanese Black cattle. BMC Genom. 2023, 24, 376. [Google Scholar] [CrossRef]
- Rubin, C.J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, X.; Li, M.; Li, Y.; Yang, Z.; Wang, X.; Pan, X.; Gong, M.; Zhang, Y.; Jiang, Y.; et al. The origin of domestication genes in goats. Sci. Adv. 2020, 6, eaaz5216. [Google Scholar] [CrossRef]
- Lee, K.D.; Millar, C.D.; Brekke, P.; Whibley, A.; Ewen, J.G.; Hingston, M.; Zhu, A.; Santure, A.W. The design and application of a 50 K SNP chip for a threatened Aotearoa New Zealand passerine, the hihi. Mol. Ecol. Resour. 2022, 22, 415–429. [Google Scholar] [CrossRef]
- Gazal, S.; Sahbatou, M.; Babron, M.C.; Génin, E.; Leutenegger, A.L. FSuite: Exploiting inbreeding in dense SNP chip and exome data. Bioinformatics 2014, 30, 1940–1941. [Google Scholar] [CrossRef] [PubMed]
- Manunza, A.; Diaz, J.R.; Sayre, B.L.; Cozzi, P.; Bobbo, T.; Deniskova, T.; Dotsev, A.; Zinovieva, N.; Stella, A. Discovering novel clues of natural selection on four worldwide goat breeds. Sci. Rep. 2023, 13, 2110. [Google Scholar] [CrossRef] [PubMed]
- Tolone, M.; Sardina, M.T.; Senczuk, G.; Chessari, G.; Criscione, A.; Moscarelli, A.; Riggio, S.; Rizzuto, I.; Di Gerlando, R.; Portolano, B.; et al. Genomic Tools for the Characterization of Local Animal Genetic Resources: Application in Mascaruna Goat. Animals 2022, 12, 2840. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Cardoso, T.F.; Marras, G.; Nicolazzi, E.L.; Rothschild, M.F.; Amills, M.; AdaptMap Consortium. Genome-wide patterns of homozygosity provide clues about the population history and adaptation of goats. Genet. Sel. Evol. 2018, 50, 59. [Google Scholar] [CrossRef] [PubMed]
- Ceccobelli, S.; Landi, V.; Senczuk, G.; Mastrangelo, S.; Sardina, M.T.; Ben-Jemaa, S.; Persichilli, C.; Karsli, T.; Balteanu, V.A.; Raschia, M.A.; et al. A comprehensive analysis of the genetic diversity and environmental adaptability in worldwide Merino and Merino-derived sheep breeds. Genet. Sel. Evol. 2023, 55, 24. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shi, L.; Li, Y.; Chen, L.; Garrick, D.; Wang, L.; Zhao, F. Estimates of genomic inbreeding and identification of candidate regions that differ between Chinese indigenous sheep breeds. J. Anim. Sci. Biotechnol. 2021, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Li, T.; Su, M.; Wang, H.; Li, Q.; Lang, X.; Ma, Y. Whole genome sequencing revealed genetic diversity, population structure, and selective signature of Panou Tibetan sheep. BMC Genom. 2023, 24, 50. [Google Scholar] [CrossRef] [PubMed]
- Szmatola, T.; Gurgul, A.; Jasielczuk, I.; Zabek, T.; Ropka-Molik, K.; Litwinczuk, Z.; Bugno-Poniewierska, M. A Comprehensive Analysis of Runs of Homozygosity of Eleven Cattle Breeds Representing Different Production Types. Animals 2019, 9, 1024. [Google Scholar] [CrossRef]
- Luo, X.; Li, J.; Xiao, C.; Sun, L.; Xiang, W.; Chen, N.; Lei, C.; Lei, H.; Long, Y.; Long, T.; et al. Whole-Genome Resequencing of Xiangxi Cattle Identifies Genomic Diversity and Selection Signatures. Front. Genet. 2022, 13, 816379. [Google Scholar] [CrossRef]
- Yin, S.; Li, Z.; Yang, F.; Guo, H.; Zhao, Q.; Zhang, Y.; Yin, Y.; Wu, X.; He, J. A Comprehensive Genomic Analysis of Chinese Indigenous Ningxiang Pigs: Genomic Breed Compositions, Runs of Homozygosity, and Beyond. Int. J. Mol. Sci. 2023, 24, 4550. [Google Scholar] [CrossRef]
- Yin, C.; Wang, Y.; Zhou, P.; Shi, H.; Ma, X.; Yin, Z.; Liu, Y. Genomic Scan for Runs of Homozygosity and Selective Signature Analysis to Identify Candidate Genes in Large White Pigs. Int. J. Mol. Sci. 2023, 24, 2914. [Google Scholar] [CrossRef] [PubMed]
- Romanov, M.N.; Abdelmanova, A.S.; Fisinin, V.I.; Gladyr, E.A.; Volkova, N.A.; Koshkina, O.A.; Rodionov, A.N.; Vetokh, A.N.; Gusev, I.V.; Anshakov, D.V.; et al. Selective footprints and genes relevant to cold adaptation and other phenotypic traits are unscrambled in the genomes of divergently selected chicken breeds. J. Anim. Sci. Biotechnol. 2023, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Tolone, M.; Sardina, M.T.; Criscione, A.; Lasagna, E.; Senczuk, G.; Rizzuto, I.; Riggio, S.; Moscarelli, A.; Macaluso, V.; Di Gerlando, R.; et al. High-density single nucleotide polymorphism markers reveal the population structure of 2 local chicken genetic resources. Poult. Sci. 2023, 102, 102692. [Google Scholar] [CrossRef] [PubMed]
- Almeida, O.A.C.; Moreira, G.C.M.; Rezende, F.M.; Boschiero, C.; de Oliveira Peixoto, J.; Ibelli, A.M.G.; Ledur, M.C.; de Novais, F.J.; Coutinho, L.L. Identification of selection signatures involved in performance traits in a paternal broiler line. BMC Genom. 2019, 20, 449. [Google Scholar] [CrossRef] [PubMed]
- Forutan, M.; Ansari Mahyari, S.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Fan, Y.; Li, W.; Yan, X.; Yan, X.; Zhang, L.; Wang, N.; Chen, O.; Zhang, Y.; Wang, R.; et al. Identification of the Key Genes Associated with Different Hair Types in the Inner Mongolia Cashmere Goat. Animals 2022, 12, 1456. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Yang, F.; Wu, Z.; Guo, F.; Zhang, J.; Hai, E.; Shang, F.; Su, R.; Wang, R.; Wang, Z.; et al. Inner Mongolian Cashmere Goat Secondary Follicle Development Regulation Research Based on mRNA-miRNA Co-analysis. Sci. Rep. 2020, 10, 4519. [Google Scholar] [CrossRef]
- Su, R.; Gong, G.; Zhang, L.; Yan, X.; Wang, F.; Zhang, L.; Qiao, X.; Li, X.; Li, J. Screening the key genes of hair follicle growth cycle in Inner Mongolian Cashmere goat based on RNA sequencing. Arch. Anim. Breed. 2020, 63, 155–164. [Google Scholar] [CrossRef]
- Dai, S.; Wang, C.; Wang, Z.; Wang, Z.; Zhang, Y.; Na, Q.; Li, J.; Wang, R. Inbreeding and its effects on fleece traits of Inner Mongolia cashmere goats. Small Rumin. Res. 2015, 128, 50–53. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, B.; Zhang, T.; Yan, X.; Yu, Y.; Li, J.; Mei, B.; Wang, Z.; Zhang, Y.; Wang, R.; et al. Assessing Genetic Diversity and Estimating the Inbreeding Effect on Economic Traits of Inner Mongolia White Cashmere Goats Through Pedigree Analysis. Front. Vet. Sci. 2021, 8, 665872. [Google Scholar] [CrossRef]
- Li, H.J. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Gorssen, W.; Meyermans, R.; Janssens, S.; Buys, N. A publicly available repository of ROH islands reveals signatures of selection in different livestock and pet species. Genet. Sel. Evol. 2021, 53, 2. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.; Ventura, R.V.; Baldi, F.; da Silva, M. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, G.; Lin, X.; Zhang, J.; Hou, G.; Zhang, L.; Liu, D.; Li, Y.; Li, J.; Xu, L. Genomic inbreeding and runs of homozygosity analysis of indigenous cattle populations in southern China. PLoS ONE 2022, 17, e0271718. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Liu, Y.; Niu, Q.; Zheng, X.; Zhang, T.; Wang, Z.; Xu, L.; Zhu, B.; Gao, X.; Zhang, L.; et al. Runs of homozygosity analysis reveals consensus homozygous regions affecting production traits in Chinese Simmental beef cattle. BMC Genom. 2021, 22, 678. [Google Scholar] [CrossRef] [PubMed]
- Ghani, M.; Sato, C.; Lee, J.H.; Reitz, C.; Moreno, D.; Mayeux, R.; St George-Hyslop, P.; Rogaeva, E. Evidence of Recessive Alzheimer Disease Loci in a Caribbean Hispanic Data Set: Genome-wide Survey of Runs of Homozygosity. JAMA Neurol. 2013, 70, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Daly, K.G.; Mattiangeli, V.; Hare, A.J.; Davoudi, H.; Fathi, H.; Doost, S.B.; Amiri, S.; Khazaeli, R.; Decruyenaere, D.; Nokandeh, J.; et al. Herded and hunted goat genomes from the dawn of domestication in the Zagros Mountains. Proc. Natl. Acad. Sci. USA 2021, 118, e2100901118. [Google Scholar] [CrossRef] [PubMed]
- Signer-Hasler, H.; Henkel, J.; Bangerter, E.; Bulut, Z.; VarGoats, C.; Drogemuller, C.; Leeb, T.; Flury, C. Runs of homozygosity in Swiss goats reveal genetic changes associated with domestication and modern selection. Genet. Sel. Evol. 2022, 54, 6. [Google Scholar] [CrossRef] [PubMed]
- Barsh, G.; Cotsarelis, G. How hair gets its pigment. Cell 2007, 130, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, W.; Liu, C.; He, Y.; Zhang, H.; Xu, L.; Yang, B.; Zhao, Y.; Ma, Y.; Chu, M.; et al. Genome-Wide Selective Analysis of Boer Goat to Investigate the Dynamic Heredity Evolution under Different Stages. Animals 2022, 12, 1356. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Hazelhurst, S.; Ramsay, M. Assessing runs of Homozygosity: A comparison of SNP Array and whole genome sequence low coverage data. BMC Genom. 2018, 19, 106. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, E.S.; Dementieva, N.V.; Shcherbakov, Y.S.; Stanishevskaya, O.I. Identification of Key Candidate Genes in Runs of Homozygosity of the Genome of Two Chicken Breeds, Associated with Cold Adaptation. Biology 2022, 11, 547. [Google Scholar] [CrossRef]
- Rostamzadeh Mahdabi, E.; Esmailizadeh, A.; Ayatollahi Mehrgardi, A.; Asadi Fozi, M. A genome-wide scan to identify signatures of selection in two Iranian indigenous chicken ecotypes. Genet. Sel. Evol. 2021, 53, 72. [Google Scholar] [CrossRef]
- Tian, S.; Tang, W.; Zhong, Z.; Wang, Z.; Xie, X.; Liu, H.; Chen, F.; Liu, J.; Han, Y.; Qin, Y.; et al. Identification of Runs of Homozygosity Islands and Functional Variants in Wenchang Chicken. Animals 2023, 13, 1645. [Google Scholar] [CrossRef] [PubMed]
- Alberto, F.J.; Boyer, F.; Orozco-terWengel, P.; Streeter, I.; Servin, B.; de Villemereuil, P.; Benjelloun, B.; Librado, P.; Biscarini, F.; Colli, L.; et al. Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 2018, 9, 813. [Google Scholar] [CrossRef] [PubMed]
- Arzik, Y.; Kizilaslan, M.; Behrem, S.; White, S.N.; Piel, L.M.W.; Cinar, M.U. Genome-Wide Scan of Wool Production Traits in Akkaraman Sheep. Genes 2023, 14, 713. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Park, J.K.; Katsnelson, J.; Kaplan, N.; Yang, W.; Getsios, S.; Lavker, R.M. microRNA-103/107 Family Regulates Multiple Epithelial Stem Cell Characteristics. Stem Cells 2015, 33, 1642–1656. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.; Chikina, M.; Clark, N. Complementary evolution of coding and noncoding sequence underlies mammalian hairlessness. eLife 2022, 11, e76911. [Google Scholar] [CrossRef]
- Li, X.; Wu, Q.; Zhang, X.; Li, C.; Zhang, D.; Li, G.; Zhang, Y.; Zhao, Y.; Shi, Z.; Wang, W.; et al. Whole-Genome Resequencing to Study Brucellosis Susceptibility in Sheep. Front. Genet. 2021, 12, 653927. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wu, P.; Chen, D.; Zhou, J.; Yang, X.; Jiang, A.; Xiao, W.; Qiu, X.; Zeng, Y.; Xu, X.; et al. Detecting the selection signatures in Chinese Duroc, Landrace, Yorkshire, Liangshan, and Qingyu pigs. Funct. Integr. Genom. 2021, 21, 655–664. [Google Scholar] [CrossRef]
- Li, X.; Kim, S.-W.; Do, K.-T.; Ha, Y.-K.; Lee, Y.-M.; Yoon, S.-H.; Kim, H.-B.; Kim, J.-J.; Choi, B.-H.; Kim, K.-S. Analyses of porcine public SNPs in coding-gene regions by re-sequencing and phenotypic association studies. Mol. Biol. Rep. 2011, 38, 3805–3820. [Google Scholar] [CrossRef]
- Gao, Y.; Gautier, M.; Ding, X.; Zhang, H.; Wang, Y.; Wang, X.; Faruque, M.O.; Li, J.; Ye, S.; Gou, X.; et al. Species composition and environmental adaptation of indigenous Chinese cattle. Sci. Rep. 2017, 7, 16196. [Google Scholar] [CrossRef]
- Na, L.; Bai, Y.; Sun, Y.; Wang, Z.; Wang, W.; Yuan, L.; Zhao, C. Identification of 9-Core Immune-Related Genes in Bladder Urothelial Carcinoma Prognosis. Front. Oncol. 2020, 10, 1142. [Google Scholar] [CrossRef]
- Nazar, M.; Abdalla, I.M.; Chen, Z.; Ullah, N.; Liang, Y.; Chu, S.; Xu, T.; Mao, Y.; Yang, Z.; Lu, X. Genome-Wide Association Study for Udder Conformation Traits in Chinese Holstein Cattle. Animals 2022, 12, 2542. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Fu, C.; Shu, S.; Wang, G.; Wang, H.; Yue, B.; Zhang, M.; Liu, X.; Liu, Y.; Zhang, J.; et al. Whole-genome resequencing of major populations revealed domestication-related genes in yaks. BMC Genom. 2024, 25, 69. [Google Scholar] [CrossRef] [PubMed]
- Braz, C.U.; Rowan, T.N.; Schnabel, R.D.; Decker, J.E. Genome-wide association analyses identify genotype-by-environment interactions of growth traits in Simmental cattle. Sci. Rep. 2021, 11, 13335. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk, E.; Vera, B.; Peraza, P.; Ciappesoni, G.; Damián, J.P.; Van Lier, E. Identification of Candidate Genes and Pathways Linked to the Temperament Trait in Sheep. Genes 2024, 15, 229. [Google Scholar] [CrossRef]
- Duan, X.-H.; Zhang, J.-H.; Huang, Y.-F.; Zhao, Y.-J.; Na, R.-S.; Zhao, Z.-Q.; Ma, Y.-H.; Chu, M.-X.; Basang, W.-D.; Zhu, Y.-B.; et al. Genome-wide selection signatures analysis of litter size in Dazu black goats using single-nucleotide polymorphism. 3 Biotech. 2019, 9, 336. [Google Scholar] [CrossRef]
- Kour, A.; Deb, S.M.; Nayee, N.; Niranjan, S.K.; Raina, V.S.; Mukherjee, A.; Patil, C.S. Novel insights into genome-wide associations in Bos indicus reveal genetic linkages between fertility and growth. Anim. Biotechnol. 2021, 34, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Lin, Z.; Song, X.; Hu, C.; Qiu, M.; Yang, L.; Liu, Y. Whole transcriptome analysis reveals the key genes and noncoding RNAs related to follicular atresia in broilers. Anim. Biotechnol. 2023, 34, 3144–3153. [Google Scholar] [CrossRef] [PubMed]
- Salilew-Wondim, D.; Hölker, M.; Rings, F.; Phatsara, C.; Mohammadi-Sangcheshmeh, A.; Tholen, E.; Schellander, K.; Tesfaye, D. Depletion of BIRC6 leads to retarded bovine early embryonic development and blastocyst formation in vitro. Reprod. Fertil. Dev. 2010, 22, 564–579. [Google Scholar] [CrossRef]
- Yao, Z.; Zhang, S.; Wang, X.; Guo, Y.; Xin, X.; Zhang, Z.; Xu, Z.; Wang, E.; Jiang, Y.; Huang, Y. Genetic diversity and signatures of selection in BoHuai goat revealed by whole-genome sequencing. BMC Genom. 2023, 24, 116. [Google Scholar] [CrossRef]
- Dixit, S.P.; Singh, S.; Ganguly, I.; Bhatia, A.K.; Sharma, A.; Kumar, N.A.; Dang, A.K.; Jayakumar, S. Genome-Wide Runs of Homozygosity Revealed Selection Signatures in Bos indicus. Front. Genet. 2020, 11, 92. [Google Scholar] [CrossRef]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scime, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Kar, D.; Ganguly, I.; Singh, S.; Bhatia, A.K.; Dixit, S.P. Genome-wide runs of homozygosity signatures in diverse Indian goat breeds. 3 Biotech. 2024, 14, 81. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Hu, M.; Shi, L.; Li, T.; Bai, C.; Sun, F.; Ma, H.; Zhao, Z.; Yan, S. Whole genome sequencing identified genomic diversity and candidated genes associated with economic traits in Northeasern Merino in China. Front. Genet. 2024, 15, 1302222. [Google Scholar] [CrossRef] [PubMed]
- Pope, M.; Budge, H.; Symonds, M.E. The developmental transition of ovine adipose tissue through early life. Acta Physiol. 2014, 210, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Saif, R.; Mahmood, T.; Zia, S.; Henkel, J.; Ejaz, A. Genomic selection pressure discovery using site-frequency spectrum and reduced local variability statistics in Pakistani Dera-Din-Panah goat. Trop. Anim. Health Prod. 2023, 55, 331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Niu, H.; Cai, Q.; Liao, M.; Chen, K.; Chen, Y.; Cong, P. Roscovitine and Trichostatin A promote DNA damage repair during porcine oocyte maturation. Reprod. Fertil. Dev. 2019, 31, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Ding, Y.; Zhang, X.; Zhou, Y.; Liu, Y.; Li, T.; Xiao, L. Whole-genome resequencing reveals new mutations in candidate genes for Beichuan-white goat prolificacya. Anim. Biotechnol. 2024, 35, 2258166. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Zhang, T.; Chen, Q.M.; Zhang, R.Q.; Li, L.; Cheng, S.F.; Shen, W.; Lei, C.Z. Genomic Signatures of Selection Associated with Litter Size Trait in Jining Gray Goat. Front. Genet. 2020, 11, 286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sui, Z.; Zhang, J.; Li, Q.; Zhang, Y.; Wang, C.; Li, X.; Xing, F. Identification of Signatures of Selection for Litter Size and Pubertal Initiation in Two Sheep Populations. Animals 2022, 12, 2520. [Google Scholar] [CrossRef]
- Martin, P.; Palhière, I.; Maroteau, C.; Clément, V.; David, I.; Klopp, G.T.; Rupp, R. Genome-wide association mapping for type and mammary health traits in French dairy goats identifies a pleiotropic region on chromosome 19 in the Saanen breed. J. Dairy Sci. 2018, 101, 5214–5226. [Google Scholar] [CrossRef]
- Lu, Q.; Gao, Y.; Fan, Z.; Xiao, X.; Chen, Y.; Si, Y.; Kong, D.; Wang, S.; Liao, M.; Chen, X.; et al. Amphiregulin promotes hair regeneration of skin-derived precursors via the PI3K and MAPK pathways. Cell Prolif. 2021, 54, e13106. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Su, R.; Wang, Z.; Zhang, Y.; Li, J. Exploring differentially expressed genes between anagen and telogen secondary hair follicle stem cells from the Cashmere goat (Capra hircus) by RNA-Seq. PLoS ONE 2020, 15, e0231376. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fan, Z.; Wang, X.; Mo, M.; Zeng, S.B.; Xu, R.-H.; Wang, X.; Wu, Y. PI3K/Akt signaling pathway is essential for de novo hair follicle regeneration. Stem Cell Res. Ther. 2020, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Lensing, M.; Jabbari, A. An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front. Immunol. 2022, 13, 955035. [Google Scholar] [CrossRef]
- Liu, Q.; Tang, Y.; Huang, Y.; Wang, J.a.; Yang, K.; Zhang, Y.; Pu, W.; Liu, J.; Shi, X.; Ma, Y.; et al. Insights into male androgenetic alopecia using comparative transcriptome profiling: Hypoxia-inducible factor-1 and Wnt/β-catenin signalling pathways. Br. J. Dermatol. 2022, 187, 936–947. [Google Scholar] [CrossRef]
- Li, C.; Feng, C.; Ma, G.; Fu, S.; Chen, M.; Zhang, W.; Li, J. Time-course RNA-seq analysis reveals stage-specific and melatonin-triggered gene expression patterns during the hair follicle growth cycle in Capra hircus. BMC Genom. 2022, 23, 140. [Google Scholar] [CrossRef]
- Ohyama, M. Use of human intra-tissue stem/progenitor cells and induced pluripotent stem cells for hair follicle regeneration. Inflamm. Regen. 2019, 39, 4. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wang, L. The potential role of ubiquitination and deubiquitination in melanogenesis. Exp. Dermatol. 2023, 32, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Kai-Yuan, J.; Yi-Wei, Z.; Ru-Jun, W.; Khan, I.M.; Yun-Hai, Z. A genome-wide integrated analysis of lncRNA-mRNA in melanocytes from white and brown skin hair boer goats (Capra aegagrus hircus). Front. Vet. Sci. 2022, 9, 1009174. [Google Scholar] [CrossRef]
- Bai, S.; Hu, S.; Dai, Y.; Jin, R.; Zhang, C.; Yao, F.; Weng, Q.; Zhai, P.; Zhao, B.; Wu, X.; et al. NRAS promotes the proliferation of melanocytes to increase melanin deposition in Rex rabbits. Genome 2022, 66, 1–10. [Google Scholar] [CrossRef]
- Hu, S.; Chen, Y.; Zhao, B.; Yang, N.; Chen, S.; Shen, J.; Bao, G.; Wu, X.J.P. KIT is involved in melanocyte proliferation, apoptosis and melanogenesis in the Rex Rabbit. PeerJ 2020, 8, e9402. [Google Scholar] [CrossRef] [PubMed]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. Genomics Confirm an Alarming Status of the Genetic Diversity of Belgian Red and Belgian White Red Cattle. Animals 2021, 11, 3574. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, W.; Wu, S.; Ma, T.; Jiang, H.; Zhang, Q. Differential expression of MC1R gene in Liaoning Cashmere goats with different coat colors. Anim. Biotechnol. 2019, 30, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.A.; Petukhova, L.; Harel, S.; Ho, Y.Y.; Drill, E.; Shapiro, L.; Wajid, M.; Christiano, A.M. FGF5 is a crucial regulator of hair length in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 10648–10653. [Google Scholar] [CrossRef] [PubMed]
- Li, W.R.; Liu, C.X.; Zhang, X.M.; Chen, L.; Peng, X.R.; He, S.G.; Lin, J.P.; Han, B.; Wang, L.Q.; Huang, J.C.; et al. CRISPR/Cas9-mediated loss of FGF5 function increases wool staple length in sheep. FEBS J. 2017, 284, 2764–2773. [Google Scholar] [CrossRef]
- Hébert, J.M.; Rosenquist, T.; Götz, J.; Martin, G.R.J.C. FGF5 as a regulator of the hair growth cycle: Evidence from targeted and spontaneous mutations. Cell 1994, 78, 1017–1025. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Breed/Population | Sample Size | Location | Use |
|---|---|---|---|---|
| IMC | Erlangshan cashmere goat | 40 | Bayannur City, Inner Mongolia, China | Cashmere and meat |
| IMC | Albas cashmere goat | 4 | Ordos City, Inner Mongolia, China | Cashmere and meat |
| IMC | Alxa cashmere goat | 7 | Alxa League, Inner Mongolia, China | Cashmere and meat |
| HSC | Hanshan White cashmere goat | 15 | Chifeng City, Inner Mongolia, China | Cashmere and meat |
| JNG | Jining Gray goat | 11 | Jining City and Heze City, Shandong Province, China | Lambskin and meat |
| ALG | Alpine goat | 10 | France | Milk |
| SAA | Saanen Dairy goat | 8 | South Korea | Milk |
| SAA | Saanen Dairy goat | 6 | The United Republic of Tanzania | Milk |
| BOE | Boer goat | 3 | South Korea | Meat |
| BOE | Boer goat | 3 | New Zealand | Meat |
| BOE | Boer goat | 6 | Australia | Meat |
| IBE | Siberian Ibex | 10 | Switzerland |
| ROH Length (Mb) | ROH Number | Number Percentage (%) | Total_Length (Mb) | Mean ± SD (Mb) | Length Percentage (%) |
|---|---|---|---|---|---|
| 0–0.3 | 25,933 | 45.32 | 6280.59 | 0.24 ± 0.028 | 25.64 |
| 0.3–1.5 | 30,406 | 53.14 | 16,313.46 | 0.54 ± 0.24 | 66.59 |
| >1.5 | 885 | 1.55 | 1905.19 | 2.15 ± 0.81 | 7.78 |
| Population | n | Total ROH Length (Mb) | Inbreeding Coefficient |
|---|---|---|---|
| IMC | 47 | 3045.20 | 0.0263 |
| HSC | 15 | 2619.12 | 0.0708 |
| JNG | 11 | 1209.02 | 0.0446 |
| SAA | 14 | 2326.06 | 0.0674 |
| ALG | 10 | 1672.71 | 0.0678 |
| BOE | 12 | 1838.68 | 0.0621 |
| IBE | 10 | 11,788.44 | 0.4780 |
| Population | Chromosome | Position (Mb) | Gene Name | Gene Function |
|---|---|---|---|---|
| ALG | 6 | 32.73~59.46 | GRID2 | Udder development, growth, fertility, temperament traits |
| 7 | 56.81~57.00 | FGF1 | Adipocyte differentiation | |
| 10 | 14.63~30.13 | TMEM63C | Body size and development | |
| MNAT1 | Cell cycle and DNA repair | |||
| BOE | 8 | 38.47~39.00 | PDCD1LG2 | Disease resistance |
| 11 | 15.04~21.07 | BIRC6 | Follicular development, fertility | |
| ARHGEF33 | Retinal development | |||
| 11 | 36.48~37.00 | ACYP2 | Growth | |
| NEBL | Puberty | |||
| 13 | 34.05~35.34 | SVIL | Disease resistance | |
| 16 | 47.76~48.17 | PRDM16 | Formation of brown fat cells, cold resistance | |
| 16 | 49.30~50.00 | CFAP74 | Meat production, staple length | |
| 19 | 22.96~23.00 | SMG6 | Growth | |
| 22 | 37.21~38.13 | PRICKLE2 | Postpartum dysgalactia syndrome | |
| HSC | 5 | 18.57~68.73 | RFX4 | Adaptability, body size and development, fertility, udder development and milk production |
| 6 | 32.54~95.45 | GRID2 | Udder development, growth, fertility, temperament traits | |
| 11 | 14.88~15.00 | BIRC6 | Follicular development, fertility | |
| 15 | 31.98~32.19 | STIM1 | Pulmonary circulation, body size and development, meat production, neural function or behavior | |
| 16 | 41.63~43.22 | RERE | Staple length | |
| 24 | 41.14~42.58 | PTPRM | Interaction between keratinocytes, meat production, immune | |
| PIEZO2 | Meat production | |||
| IMC | 1 | 82.57~108.78 | ECE2 | Fertility, embryo development |
| 6 | 95.44~116.63 | FGF5 | Hair growth and length | |
| SH3BP2 | Immune | |||
| 11 | 14.98~15.04 | BIRC6 | Follicular development, fertility | |
| 15 | 31.97~32.19 | STIM1 | Pulmonary circulation, body size and development, meat production, neural function or behavior | |
| 19 | SMG6 | Growth | ||
| JNG | 1 | 109.15~132.19 | STAG1 | Embryonic development |
| SAA | 5 | 59.27~69.98 | SYN3 | Body size and development, meat production |
| 5 | 70.00~98.96 | SYN3 | Body size and development, meat production | |
| 7 | 27.59~57.47 | XRCC4 | Embryonic development | |
| 7 | 58.31~59.89 | MATR3 | Corpus luteum | |
| 8 | 74.99~75.00 | NOL6 | Skin color | |
| 11 | 78.35~93.90 | SDC1 | Body size and development, meat production, cashmere fineness | |
| STRBP | Body size | |||
| 19 | 24.51~40.23 | RARA | Milk production, udder development, hair follicle morphogenesis | |
| 28 | CDH23 | Fertility | ||
| 29 | 17.94~18.00 | PAK1 | Fertility, puberty |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Q.; Huang, C.; Chen, Q.; Su, Y.; Zhang, Y.; Wang, R.; Su, R.; Xu, H.; Liu, S.; Ma, Y.; et al. Genomic Inbreeding and Runs of Homozygosity Analysis of Cashmere Goat. Animals 2024, 14, 1246. https://doi.org/10.3390/ani14081246
Zhao Q, Huang C, Chen Q, Su Y, Zhang Y, Wang R, Su R, Xu H, Liu S, Ma Y, et al. Genomic Inbreeding and Runs of Homozygosity Analysis of Cashmere Goat. Animals. 2024; 14(8):1246. https://doi.org/10.3390/ani14081246
Chicago/Turabian StyleZhao, Qian, Chang Huang, Qian Chen, Yingxiao Su, Yanjun Zhang, Ruijun Wang, Rui Su, Huijuan Xu, Shucai Liu, Yuehui Ma, and et al. 2024. "Genomic Inbreeding and Runs of Homozygosity Analysis of Cashmere Goat" Animals 14, no. 8: 1246. https://doi.org/10.3390/ani14081246
APA StyleZhao, Q., Huang, C., Chen, Q., Su, Y., Zhang, Y., Wang, R., Su, R., Xu, H., Liu, S., Ma, Y., Zhao, Q., & Ye, S. (2024). Genomic Inbreeding and Runs of Homozygosity Analysis of Cashmere Goat. Animals, 14(8), 1246. https://doi.org/10.3390/ani14081246