
Review of Molecular Technologies for Investigating Canine Cancer



Abstract
Simple Summary
Abstract
1. Introduction
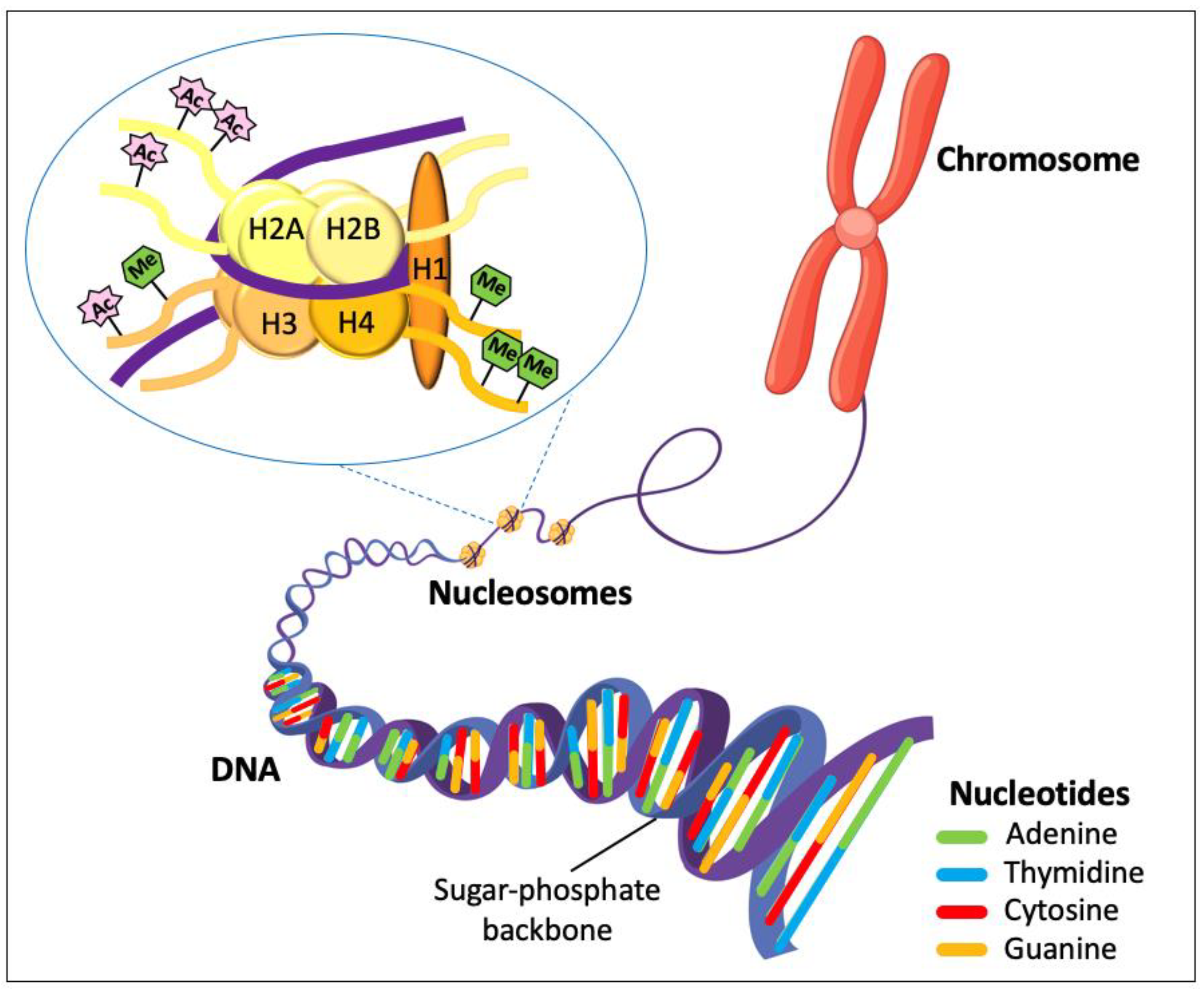
1.1. The Genome and Genetic Alterations
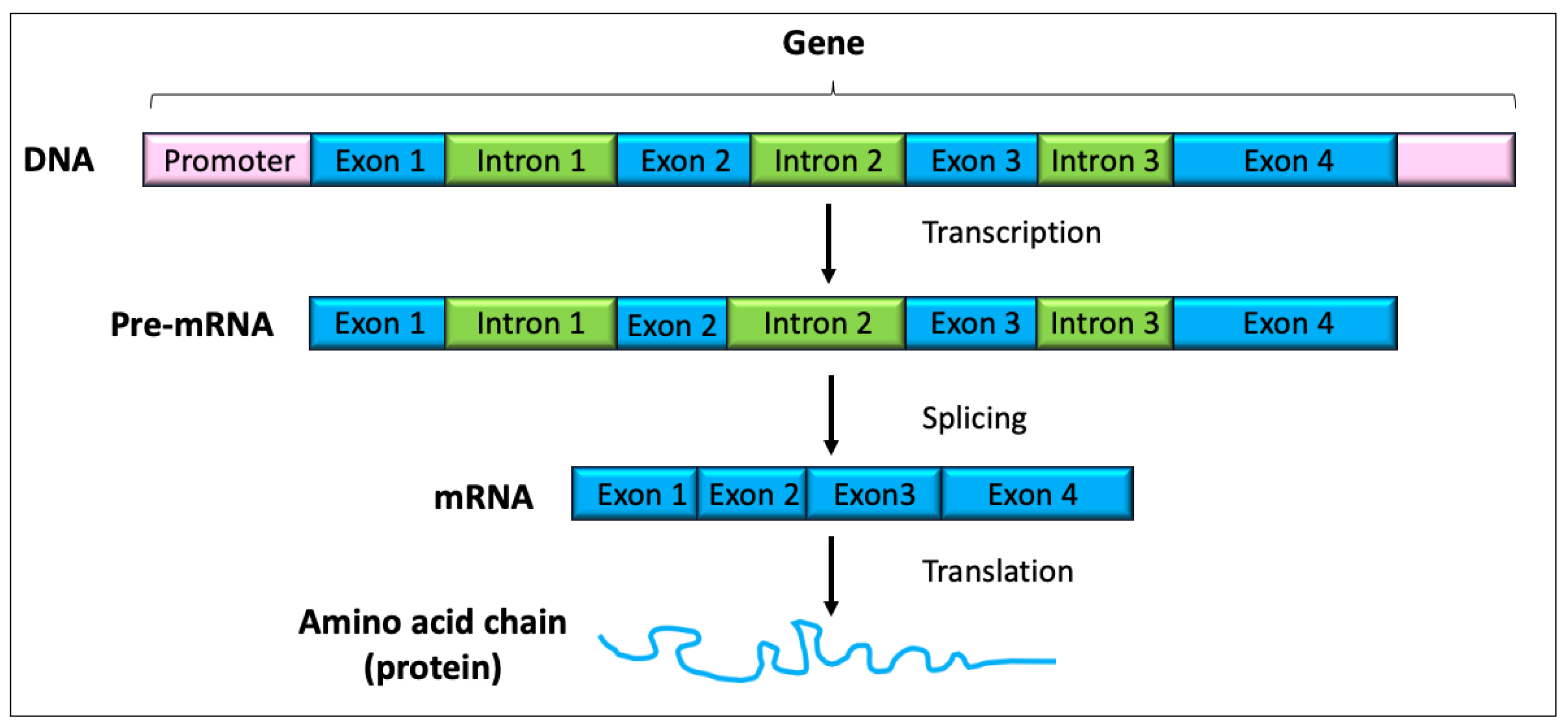
1.2. The Transcriptome and Alterations in Gene Expression
1.3. The Epigenome and Epigenetic Alterations
1.4. Oncogenomics
2. Methods for Sampling Tumour Cells in Dogs
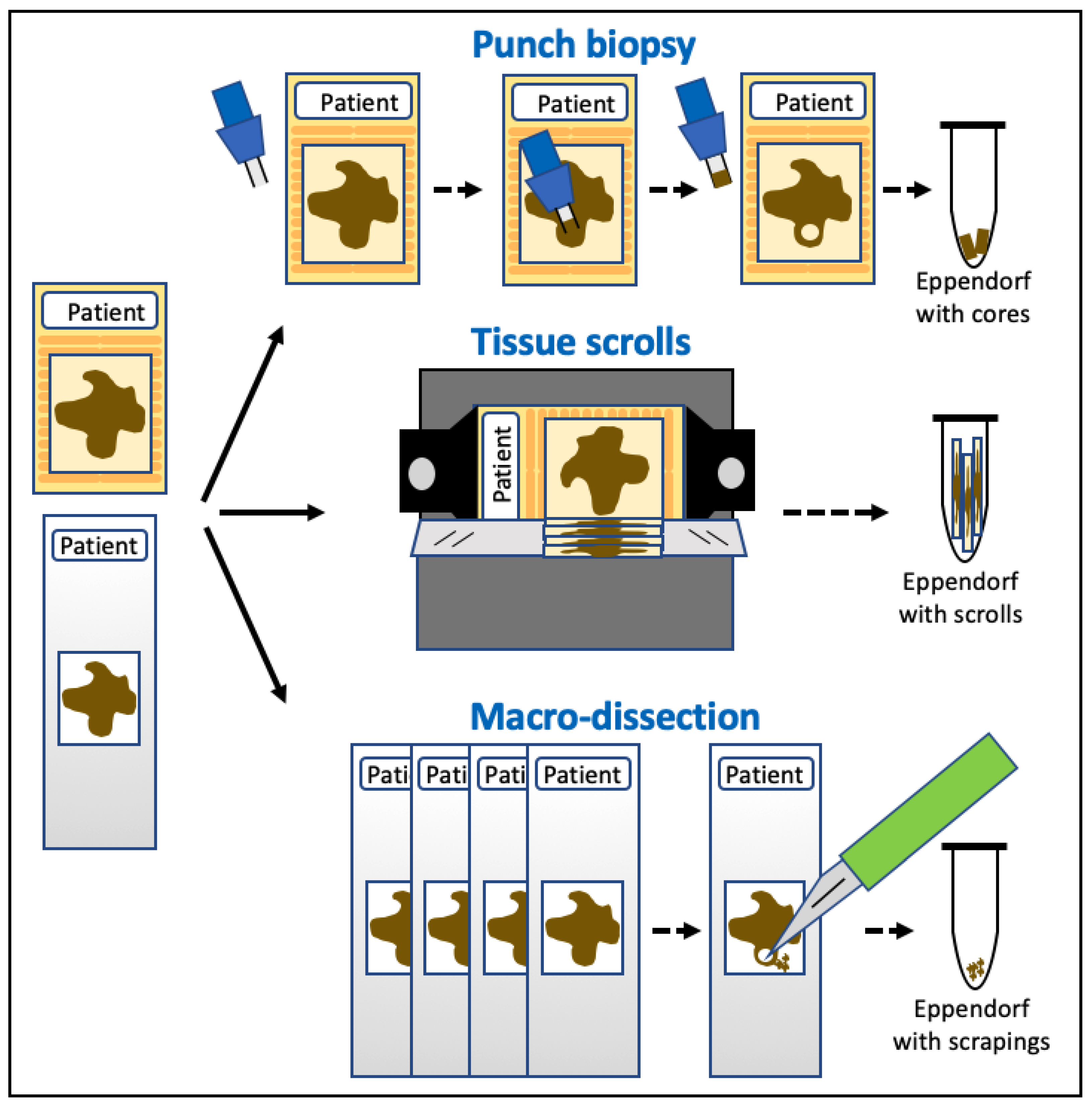
2.1. Tissue Samples
2.2. Liquid Biopsy
2.2.1. Circulating Tumour Cells
2.2.2. Circulating Cell-Free DNA
2.2.3. Nucleosomes
2.2.4. MicroRNAs
3. Methods for the Genetic Characterisation of Tumour Cells in Dogs
3.1. Polymerase Chain Reaction (PCR)
3.1.1. PCR Using DNA
3.1.2. PCR Using RNA
3.1.3. Quantitative PCR (qPCR)
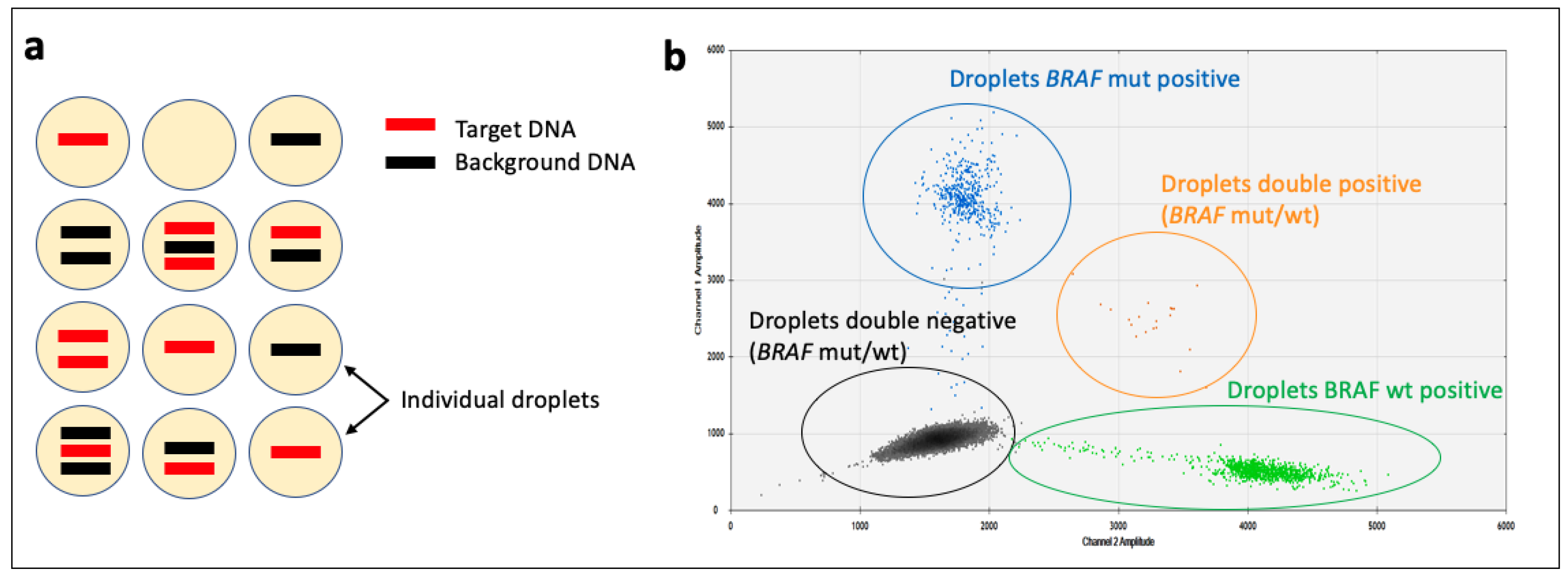
3.1.4. Digital PCR (dPCR)
3.2. DNA Microarray
3.2.1. Gene Expression Arrays
3.2.2. Array-Based Comparative Genomic Hybridization (aCGH)
3.2.3. Microarray-Based DNA Methylation Profiling
3.3. Quantitative Nuclease Protection Assay (qNPA)
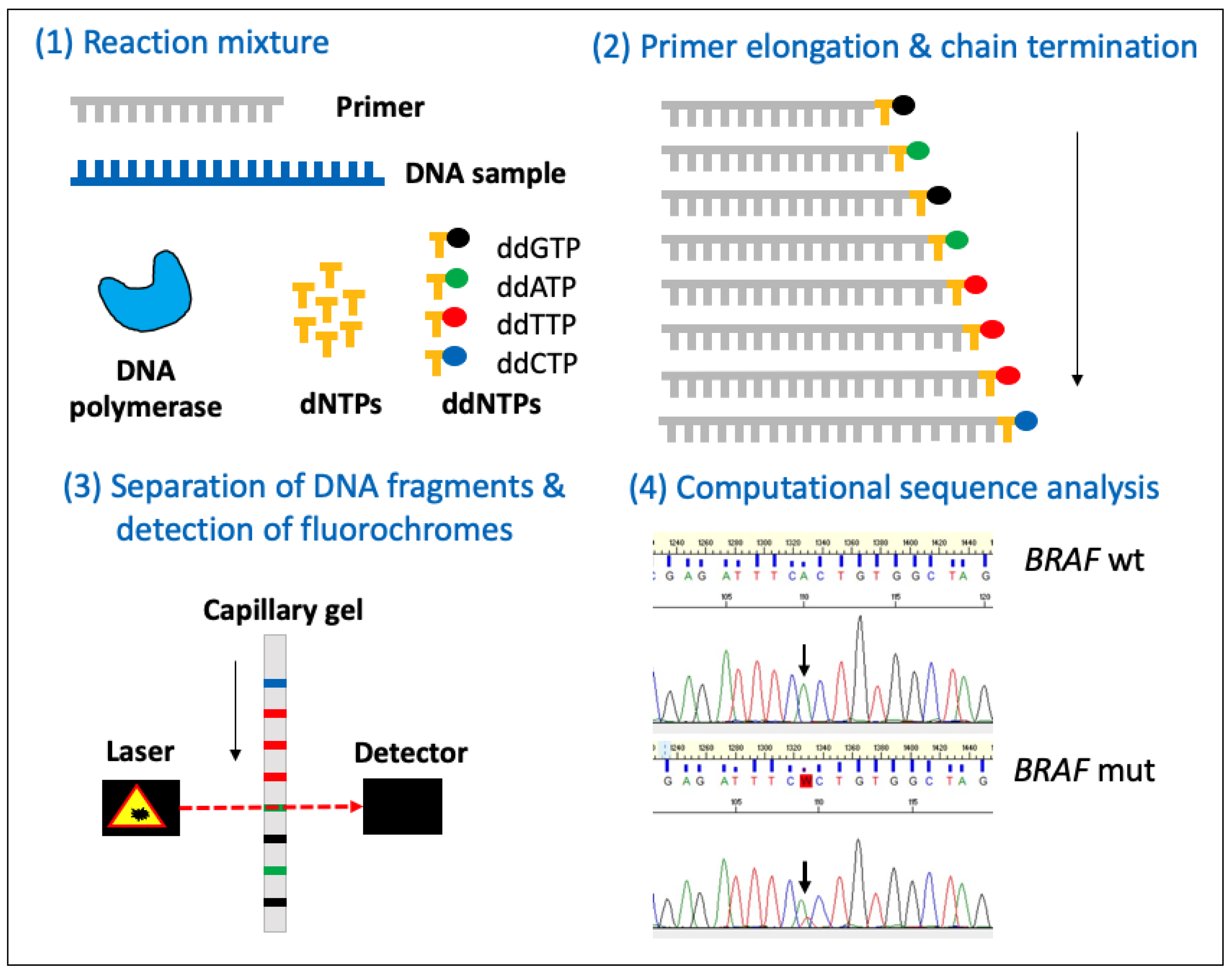
3.4. Sanger Sequencing
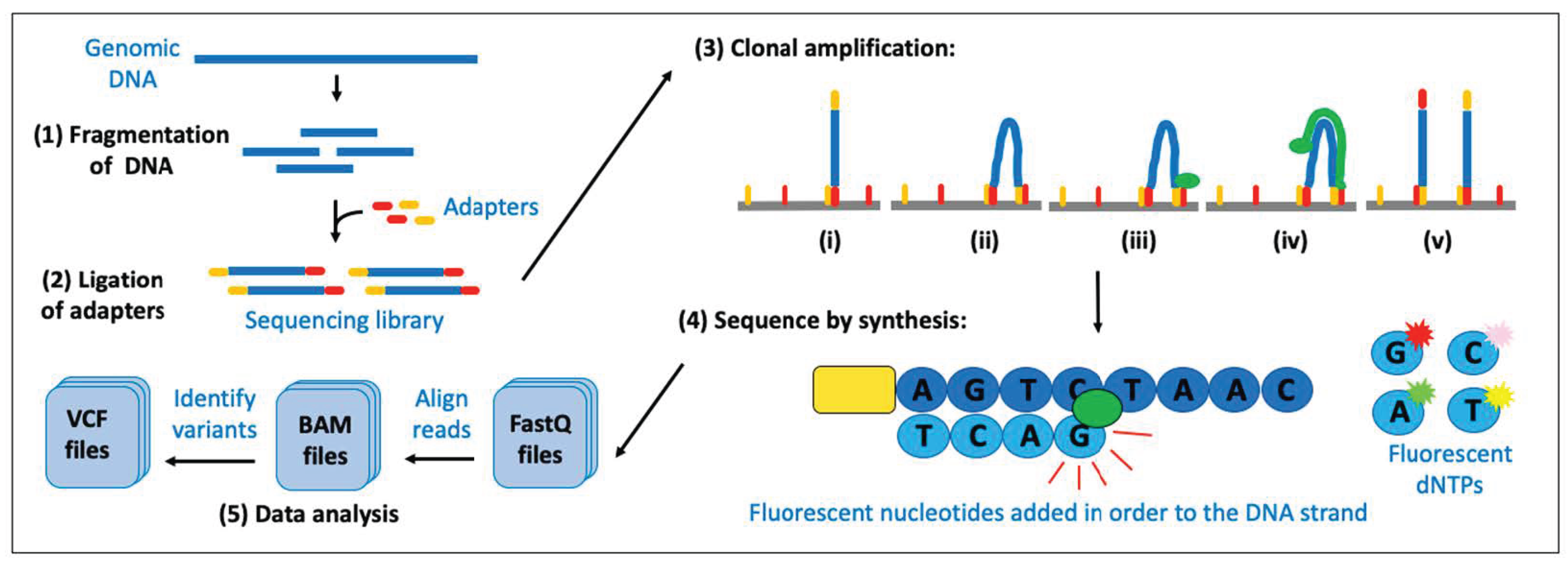
3.5. Next-Generation Sequencing (NGS)
3.5.1. NGS for Detecting Variations in the DNA
- (1)
- A sequence that is complementary to the solid support. The solid support comprises the oligonucleotides that are covalently attached to the surface of the flow cell of the sequencing machine and are required for the ‘cluster amplification’ step later in the procedure.
- (2)
- A ‘barcode’ sequence. This is a short unique tag. All fragments of DNA from one sample (library) have adapters containing the same tag, to allow for multiple libraries to be mixed together and sequenced at the same time (known as ‘multiplexing’ or ‘pooling’).
- (3)
- A binding site for the sequencing primer. This is required for the ‘sequencing’ step later in the procedure.
3.5.2. NGS for Detecting Variations in the RNA
3.5.3. Methylation Sequencing
3.6. A Reference Genome for Domestic Dogs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Name | Breed (Sex); Name | Accession ID | Ref |
|---|---|---|---|
| CanFam3.1 | Boxer (F); ‘Tasha’ | GCA_000002285.2 | [101] |
| ROS_Cfam_1.0 | Labrador Retriever (M) | GCA_014441545.1 | TRI, 2020 |
| ASM864105v3 (synonym: CanFam_GSD) | German Shepherd (F); ‘Nala’ | GCA_008641055.3 | [109] |
| ASM1204501v1 | Labrador Retriever (M); ‘Yella’ | GCA_012045015.1 | [110] |
| CanFam_Bas | Basenji (F); ‘China’ Basenji (M); ‘Wags’ | GCA_013276365.1 GCA_013276365.2 | [111] |
| UU_Cfam_GSD_1.0 (synonym: CanFam4) | German Shepherd (F); ‘Mischka’ | GCA_011100685.1 | [103] |
| UMICH_Zoey_3.1 (Synonym: CanFam5) | Great Dane (F); ‘Zoey’ | GCA_005444595.1 | [112] |
| Dog10K_Boxer_Tasha_1.0 (synonym: CanFam6) | ‘Tasha’ (details as above) | GCA_000002285.4 | [104] |
| CA611_1.0 | Cairn Terrier (M) | GCA_031010295.1 | [113] |
| BD_1.0 | Bernese Mountain Dog (F) | GCA_031010765.1 | [113] |
| OD_1.0 | Bernese Mountain Dog (M) | GCA_031010635.1 | [113] |
3.7. Germline Databases
4. Emerging Fields for Genetic Investigations of Canine Tumours
4.1. RNA Analysis
4.1.1. Single-Cell RNA Sequencing (scRNA-Seq)
4.1.2. Spatial Transcriptomics (ST)
- (1)
- ISH methods (such as seqFISH, merFISH and seqFISH+) detect specific target genes through the use of fluorescently labelled probes that are complementary to the RNA transcript of interest, with the signals from the probes providing quantitative determination of the transcripts in that spatial context.
- (2)
- ISS methods (such as Padlock Probe ISS and FISSEQ) involve fixation of the mRNA, followed by in situ reverse transcription to form cDNA. Padlock probes (PLPs; single-stranded DNA probes designed against targets of interest) are then hybridised with the tissue section and allowed to bind with the cDNA. Bound PLPs are amplified by a process known as rolling circle amplification (RCA) and are labelled with fluorophore-conjugated probes, which allow their detection. ISS can detect up to a few hundred genes per sample and wide-field imaging enables high throughput.
- (3)
- ISC methods (such as 10× Genomics Visium, Slide-seq and Seq-Scope) capture transcripts in situ, whilst NGS is performed ex situ and, as such, enables unbiased capture of the entire transcriptome. The general ISC strategy uses slides with arrays of ‘capture spots’ consisting of barcoded reverse transcription (RT) primers with poly-T sequences that capture the mRNA transcripts. Tissues are sectioned onto these slides to allow hybridisation of the transcripts to the spots, after which RT is performed, and the resulting cDNAs are extracted for NGS. After sequencing, the reads are superimposed onto the tissue image using the positional barcodes, thus allowing spatial visualisation of the transcriptome.
4.2. Prediction of Genetic Alterations from Histology Slides
5. Important Considerations
5.1. Adaptation of Technologies Used in Human Research
5.2. Generation of Databases for Our Genomic Knowledge
- Cancer Genome Atlas (TCGA): collected, characterised, and analysed human cancer samples from >11,000 patients over a 12-year period (https://www.cancer.gov/ccg/research/genome-sequencing/tcga)
- Catalogue Of Somatic Mutations In Cancer (COSMIC): the world’s largest and most comprehensive resource for exploring the impact of somatic mutations in human cancer (https://cancer.sanger.ac.uk/cosmic)
- cBioPortal for Cancer Genomics: provides visualisation, analysis and a download of large-scale human cancer genomics data sets (https://www.cbioportal.org/)
5.3. Ethics
6. Conclusions
- (1)
- The nature of the investigation; for example, does it require analysing the whole genome or just a portion of the genome, an entire gene or just a specific portion of a gene? Is single-cell and/or spatial analysis required, or will bulk analysis of the tumour suffice?
- (2)
- Availability of access to the relevant equipment required; for example, does the method need NGS technology or will a PCR machine suffice?
- (3)
- Complexity of the data output; for example, does it require a bioinformatician to analyse the sequencing data, or does it not require any (or only minimal) computational analysis?
- (4)
- Time frame; for example, an NGS and the subsequent data analysis can take several months.
- (5)
- Cost; for example, WGS costs more than WES, and RNA-Seq costs more than qPCR.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Estabrooks, T.; Gurinovich, A.; Pietruska, J.; Lewis, B.; Harvey, G.; Post, G.; Lambert, L.; Miller, A.; Rodrigues, L.; White, M.E. Identification of genomic alterations with clinical impact in canine splenic hemangiosarcoma. Vet. Comp. Oncol. 2022, 21, 623–633. [Google Scholar] [CrossRef]
- Sakthikumar, S.; Warrier, M.; Whitley, D.; Facista, S.; Adkins, J.; Aman, S.; Tsinajinnie, D.; Duran, N.; Siravegna, G.; Ahmed, Z.; et al. Genomic analysis across 53 canine cancer types reveals novel mutations and high clinical actionability potential. Vet. Comp. Oncol. 2023, 22, 30–41. [Google Scholar] [CrossRef]
- Chon, E.; Sakthikumar, S.; Tang, M.; Hamilton, M.J.; Vaughan, A.; Smith, A.; Sommer, B.; Robat, C.; Manley, C.; Mullin, C.; et al. Novel genomic prognostic biomarkers for dogs with cancer. J. Vet. Intern. Med. 2023, 37, 2410–2421. [Google Scholar] [CrossRef]
- Wu, K.; Rodrigues, L.; Post, G.; Harvey, G.; White, M.; Miller, A.; Lambert, L.; Lewis, B.; Lopes, C.; Zou, J. Analyses of canine cancer mutations and treatment outcomes using real-world clinico-genomics data of 2119 dogs. NPJ Precis. Oncol. 2023, 7, 8. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Alberti, M.O.; Au, C.; Li, Y.; Toy, T.; Tomasian, V.; Xian, R.R. Nucleic Acid Extraction from Human Biological Samples. Methods Mol. Biol. 2019, 1897, 359–383. [Google Scholar] [CrossRef] [PubMed]
- Sailasuta, A.; Ketpun, D.; Piyaviriyakul, P.; Theerawatanasirikul, S.; Theewasutrakul, P.; Rungsipipat, A. The Relevance of CD117-Immunocytochemistry Staining Patterns to Mutational Exon-11 in c-kit Detected by PCR from Fine-Needle Aspirated Canine Mast Cell Tumor Cells. Vet. Med. Int. 2014, 2014, 787498. [Google Scholar] [CrossRef] [PubMed]
- Mathieson, W.; Thomas, G.A. Why Formalin-fixed, Paraffin-embedded Biospecimens Must Be Used in Genomic Medicine: An Evidence-based Review and Conclusion. J. Histochem. Cytochem. 2020, 68, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Steiert, T.A.; Parra, G.; Gut, M.; Arnold, N.; Trotta, J.R.; Tonda, R.; Moussy, A.; Gerber, Z.; Abuja, P.M.; Zatloukal, K.; et al. A critical spotlight on the paradigms of FFPE-DNA sequencing. Nucleic Acids Res. 2023, 51, 7143–7162. [Google Scholar] [CrossRef]
- Newton, Y.; Sedgewick, A.J.; Cisneros, L.; Golovato, J.; Johnson, M.; Szeto, C.W.; Rabizadeh, S.; Sanborn, J.Z.; Benz, S.C.; Vaske, C. Large scale, robust, and accurate whole transcriptome profiling from clinical formalin-fixed paraffin-embedded samples. Sci. Rep. 2020, 10, 17597. [Google Scholar] [CrossRef] [PubMed]
- Groelz, D.; Viertler, C.; Pabst, D.; Dettmann, N.; Zatloukal, K. Impact of storage conditions on the quality of nucleic acids in paraffin embedded tissues. PLoS ONE 2018, 13, e0203608. [Google Scholar] [CrossRef] [PubMed]
- Tamlin, V.S.; Dobson, E.C.; Woolford, L.; Peaston, A.E. DNA purification increases PCR-amplifiable DNA extracted from formalin-fixed, paraffin-embedded canine mast cell tumors for routine KIT mutation detection. J. Vet. Diagn. Investig. 2019, 31, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Marczyk, M.; Fu, C.; Lau, R.; Du, L.; Trevarton, A.J.; Sinn, B.V.; Gould, R.E.; Pusztai, L.; Hatzis, C.; Symmans, W.F. The impact of RNA extraction method on accurate RNA sequencing from formalin-fixed paraffin-embedded tissues. BMC Cancer 2019, 19, 1189. [Google Scholar] [CrossRef]
- Kofanova, O.; Bellora, C.; Garcia Frasquilho, S.; Antunes, L.; Hamot, G.; Mathay, C.; Mommaerts, K.; Muller, A.; DeWitt, B.; Betsou, F. Standardization of the preanalytical phase of DNA extraction from fixed tissue for next-generation sequencing analyses. N. Biotechnol. 2020, 54, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Barreira da Costa, A.; Kohn, B.; Oliveira, J.; Gärtner, F.; Gruber, A.D.; Klopfleisch, R. Genetic Markers for the Detection of Circulating Tumour Cells in Dogs with Metastatic Mammary Tumours. J. Comp. Pathol. 2010, 143, 314. [Google Scholar] [CrossRef]
- da Costa, A.; Lenze, D.; Hummel, M.; Kohn, B.; Gruber, A.D.; Klopfleisch, R. Identification of six potential markers for the detection of circulating canine mammary tumour cells in the peripheral blood identified by microarray analysis. J. Comp. Pathol. 2012, 146, 143–151. [Google Scholar] [CrossRef]
- da Costa, A.; Kohn, B.; Gruber, A.D.; Klopfleisch, R. Multiple RT-PCR markers for the detection of circulating tumour cells of metastatic canine mammary tumours. Vet. J. 2013, 196, 34–39. [Google Scholar] [CrossRef]
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Beffagna, G.; Sammarco, A.; Bedin, C.; Romualdi, C.; Mainenti, M.; Mollo, A.; Cavicchioli, L.; Ferro, S.; Trez, D.; De Maria, R.; et al. Circulating Cell-Free DNA in Dogs with Mammary Tumors: Short and Long Fragments and Integrity Index. PLoS ONE 2017, 12, e0169454. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, D.M.; Forman, M.A.; Kisseberth, W.C.; Lehman, A.M.; Kelbick, N.T.; Harper, P.; Rush, L.J. Quantification of plasma DNA as a prognostic indicator in canine lymphoid neoplasia. Vet. Comp. Oncol. 2007, 5, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Prouteau, A.; Denis, J.A.; De Fornel, P.; Cadieu, E.; Derrien, T.; Kergal, C.; Botherel, N.; Ulvé, R.; Rault, M.; Bouzidi, A.; et al. Circulating tumor DNA is detectable in canine histiocytic sarcoma, oral malignant melanoma, and multicentric lymphoma. Sci. Rep. 2021, 11, 877. [Google Scholar] [CrossRef] [PubMed]
- Flory, A.; Kruglyak, K.M.; Tynan, J.A.; McLennan, L.M.; Rafalko, J.M.; Fiaux, P.C.; Hernandez, G.E.; Marass, F.; Nakashe, P.; Ruiz-Perez, C.A.; et al. Clinical validation of a next-generation sequencing-based multi-cancer early detection “liquid biopsy” blood test in over 1,000 dogs using an independent testing set: The CANcer Detection in Dogs (CANDiD) study. PLoS ONE 2022, 17, e0266623. [Google Scholar] [CrossRef] [PubMed]
- O’Kell, A.L.; Lytle, K.M.; Cohen, T.A.; Wong, L.K.; Sandford, E.; Rafalko, J.M.; Brandstetter, G.; DiMarzio, L.R.; Phelps-Dunn, A.; Rosentel, M.C.; et al. Clinical experience with next-generation sequencing-based liquid biopsy testing for cancer detection in dogs: A review of 1,500 consecutive clinical cases. J. Am. Vet. Med. Assoc. 2023, 261, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Abascal, F.; Ludwig, L.; Aupperle-Lellbach, H.; Grassinger, J.; Wright, C.W.; Allison, S.J.; Pinder, E.; Phillips, R.M.; Romero, L.P.; et al. Cross-species oncogenomics offers insight into human muscle-invasive bladder cancer. Genome Biol. 2023, 24, 191. [Google Scholar] [CrossRef] [PubMed]
- Decker, B.; Parker, H.G.; Dhawan, D.; Kwon, E.M.; Karlins, E.; Davis, B.W.; Ramos-Vara, J.A.; Bonney, P.L.; McNiel, E.A.; Knapp, D.W.; et al. Homologous Mutation to Human BRAF V600E Is Common in Naturally Occurring Canine Bladder Cancer--Evidence for a Relevant Model System and Urine-Based Diagnostic Test. Mol. Cancer Res. 2015, 13, 993–1002. [Google Scholar] [CrossRef]
- Aupperle-Lellbach, H.; Grassinger, J.; Hohloch, C.; Kehl, A.; Pantke, P. Diagnostic value of the BRAF variant V595E in urine samples, smears and biopsies from canine transitional cell carcinoma. Tierarztl. Praxis. Ausg. K Kleintiere Heimtiere 2018, 46, 289–295. [Google Scholar] [CrossRef]
- Fedyuk, V.; Erez, N.; Furth, N.; Beresh, O.; Andreishcheva, E.; Shinde, A.; Jones, D.; Zakai, B.B.; Mavor, Y.; Peretz, T.; et al. Multiplexed, single-molecule, epigenetic analysis of plasma-isolated nucleosomes for cancer diagnostics. Nat. Biotechnol. 2023, 41, 212–221. [Google Scholar] [CrossRef]
- Gezer, U.; Yörüker, E.E.; Keskin, M.; Kulle, C.B.; Dharuman, Y.; Holdenrieder, S. Histone Methylation Marks on Circulating Nucleosomes as Novel Blood-Based Biomarker in Colorectal Cancer. Int. J. Mol. Sci. 2015, 16, 29654–29662. [Google Scholar] [CrossRef]
- McAnena, P.; Brown, J.A.; Kerin, M.J. Circulating Nucleosomes and Nucleosome Modifications as Biomarkers in Cancer. Cancers 2017, 9, 5. [Google Scholar] [CrossRef]
- Dolan, C.; Miller, T.; Jill, J.; Terrell, J.; Kelly, T.K.; Bygott, T.; Wilson-Robles, H. Characterizing circulating nucleosomes in the plasma of dogs with lymphoma. BMC Vet. Res. 2021, 17, 276. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Robles, H.M.; Bygott, T.; Kelly, T.K.; Miller, T.M.; Miller, P.; Matsushita, M.; Terrell, J.; Bougoussa, M.; Butera, T. Evaluation of plasma nucleosome concentrations in dogs with a variety of common cancers and in healthy dogs. BMC Vet. Res. 2022, 18, 329. [Google Scholar] [CrossRef] [PubMed]
- Letendre, J.A.; Goggs, R. Concentrations of Plasma Nucleosomes but Not Cell-Free DNA Are Prognostic in Dogs Following Trauma. Front. Vet. Sci. 2018, 5, 180. [Google Scholar] [CrossRef] [PubMed]
- Letendre, J.A.; Goggs, R. Determining prognosis in canine sepsis by bedside measurement of cell-free DNA and nucleosomes. J. Vet. Emerg. Crit. Care 2018, 28, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.K.H.; Lyngby, J.G.; Bjørnvad, C.R.; Nielsen, L.N. Presence of nucleosomes in plasma and increased thrombin generation in dogs with acute and chronic gastroenteropathies. Res. Vet. Sci. 2021, 135, 504–510. [Google Scholar] [CrossRef]
- Syed, S.N.; Brüne, B. Exosomal and Non-Exosomal MicroRNAs: New Kids on the Block for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 4493. [Google Scholar] [CrossRef]
- Sohel, M.M.H. Circulating microRNAs as biomarkers in cancer diagnosis. Life Sci. 2020, 248, 117473. [Google Scholar] [CrossRef]
- Sempere, L.F.; Zaluzec, E.; Kenyon, E.; Kiupel, M.; Moore, A. Automated Five-Color Multiplex Co-detection of MicroRNA and Protein Expression in Fixed Tissue Specimens. Methods Mol. Biol. 2020, 2148, 257–276. [Google Scholar] [CrossRef]
- Chakraborty, S.; Banerjee, S. Multidimensional computational study to understand non-coding RNA interactions in breast cancer metastasis. Sci. Rep. 2023, 13, 15771. [Google Scholar] [CrossRef]
- Heishima, K.; Mori, T.; Ichikawa, Y.; Sakai, H.; Kuranaga, Y.; Nakagawa, T.; Tanaka, Y.; Okamura, Y.; Masuzawa, M.; Sugito, N.; et al. MicroRNA-214 and MicroRNA-126 Are Potential Biomarkers for Malignant Endothelial Proliferative Diseases. Int. J. Mol. Sci. 2015, 16, 25377–25391. [Google Scholar] [CrossRef]
- Heishima, K.; Meuten, T.; Yoshida, K.; Mori, T.; Thamm, D.H. Prognostic significance of circulating microRNA-214 and -126 in dogs with appendicular osteosarcoma receiving amputation and chemotherapy. BMC Vet. Res. 2019, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- Lyngby, J.G.; Gòdia, M.; Brogaard, L.; Kristensen, A.T.; Fredholm, M.; Skancke, E.; Morris, J.; Dupont, N.; Salavati Schmitz, S.; Argyle, D.; et al. Association of fecal and serum microRNA profiles with gastrointestinal cancer and chronic inflammatory enteropathy in dogs. J. Vet. Intern. Med. 2022, 36, 1989–2001. [Google Scholar] [CrossRef]
- Fish, E.J.; Martinez-Romero, E.G.; DeInnocentes, P.; Koehler, J.W.; Prasad, N.; Smith, A.N.; Bird, R.C. Circulating microRNA as biomarkers of canine mammary carcinoma in dogs. J. Vet. Intern. Med. 2020, 34, 1282–1290. [Google Scholar] [CrossRef]
- Jain, M.; Ingole, S.D.; Deshmukh, R.S.; Bharucha, S.V.; Nagvekar, A.S.; Gaikwad, R.V.; Kharde, S.D. CEA, CA 15-3, and miRNA expression as potential biomarkers in canine mammary tumors. Chromosome Res. 2021, 29, 175–188. [Google Scholar] [CrossRef]
- Ramadan, E.S.; Salem, N.Y.; Emam, I.A.; AbdElKader, N.A.; Farghali, H.A.; Khattab, M.S. MicroRNA-21 expression, serum tumor markers, and immunohistochemistry in canine mammary tumors. Vet. Res. Commun. 2022, 46, 377–388. [Google Scholar] [CrossRef]
- Garnica, T.K.; Lesbon, J.C.C.; Ávila, A.; Rochetti, A.L.; Matiz, O.R.S.; Ribeiro, R.C.S.; Zoppa, A.; Nishiya, A.T.; Costa, M.T.; de Nardi, A.B.; et al. Liquid biopsy based on small extracellular vesicles predicts chemotherapy response of canine multicentric lymphomas. Sci. Rep. 2020, 10, 20371. [Google Scholar] [CrossRef]
- Fujiwara-Igarashi, A.; Igarashi, H.; Mizutani, N.; Goto-Koshino, Y.; Takahashi, M.; Ohno, K.; Tsujimoto, H. Expression profile of circulating serum microRNAs in dogs with lymphoma. Vet. J. 2015, 205, 317–321. [Google Scholar] [CrossRef]
- Vinall, R.L.; Kent, M.S.; deVere White, R.W. Expression of microRNAs in urinary bladder samples obtained from dogs with grossly normal bladders, inflammatory bladder disease, or transitional cell carcinoma. Am. J. Vet. Res. 2012, 73, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Kent, M.S.; Zwingenberger, A.; Westropp, J.L.; Barrett, L.E.; Durbin-Johnson, B.P.; Ghosh, P.; Vinall, R.L. MicroRNA profiling of dogs with transitional cell carcinoma of the bladder using blood and urine samples. BMC Vet. Res. 2017, 13, 339. [Google Scholar] [CrossRef]
- Rivera, P.L.; Li, W.T.; Bhogal, S.; Mandell, J.B.; Belayneh, R.; Hankins, M.L.; Payne, J.T.; Watters, R.J.; Weiss, K.R. Antioxidant 1 copper chaperone gene expression and copper levels in dog osteosarcoma patients. Vet. Comp. Oncol. 2023, 21, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.; Hicks, J.L.; De Marzo, A.M.; Campbell, A.A.; Eberhart, C.G.; Dubielzig, R.R.; Teixeira, L.B. Upregulated MYC expression and p53 mutations may contribute to the oncogenesis of canine Meibomian gland carcinomas. Vet. Pathol. 2023, 60, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Klopfleisch, R.; Lenze, D.; Hummel, M.; Gruber, A.D. Metastatic canine mammary carcinomas can be identified by a gene expression profile that partly overlaps with human breast cancer profiles. BMC Cancer 2010, 10, 618. [Google Scholar] [CrossRef]
- Seitz, J.; Bilsland, A.; Puget, C.; Baasner, I.; Klopfleisch, R.; Stein, T. SFRP1 Expression is Inversely Associated With Metastasis Formation in Canine Mammary Tumours. J. Mammary Gland. Biol. Neoplasia 2023, 28, 15. [Google Scholar] [CrossRef] [PubMed]
- Ettlin, J.; Bauer, A.; Opitz, L.; Malbon, A.; Markkanen, E. Deciphering Stromal Changes between Metastatic and Non-metastatic Canine Mammary Carcinomas. J. Mammary Gland. Biol. Neoplasia 2023, 28, 14. [Google Scholar] [CrossRef] [PubMed]
- Irving, J.R.; Hiron, T.K.; Davison, L.J.; Xia, D.; Beck, S.; Werling, D.; Williams, J. Characterization of canine intestinal microRNA expression in inflammatory bowel disease and T-cell lymphoma. J. Comp. Pathol. 2023, 204, 23–29. [Google Scholar] [CrossRef]
- Brunstein, J. Digital PCR: Theory and applications. Med. Lab. Obs. 2013, 45, 34–35. [Google Scholar]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Shapiro, S.G.; Breen, M. Detection of BRAF Mutation in Urine DNA as a Molecular Diagnostic for Canine Urothelial and Prostatic Carcinoma. PLoS ONE 2015, 10, e0144170. [Google Scholar] [CrossRef]
- Conrad, D.; Kehl, A.; Beitzinger, C.; Metzler, T.; Steiger, K.; Pfarr, N.; Fischer, K.; Klopfleisch, R.; Aupperle-Lellbach, H. Molecular Genetic Investigation of Digital Melanoma in Dogs. Vet. Sci. 2022, 9, 56. [Google Scholar] [CrossRef]
- Asada, H.; Tomiyasu, H.; Okada, K.; Chambers, J.K.; Goto-Koshino, Y.; Uchida, K.; Kagawa, Y.; Ohno, K.; Tsujimoto, H. Clinical significance of the two-base insertion mutation in the TP53 gene in canine histiocytic sarcoma. Res. Vet. Sci. 2019, 124, 57–60. [Google Scholar] [CrossRef]
- Whale, A.S.; Huggett, J.F.; Cowen, S.; Speirs, V.; Shaw, J.; Ellison, S.; Foy, C.A.; Scott, D.J. Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res. 2012, 40, e82. [Google Scholar] [CrossRef]
- Mochizuki, H.; Thomas, R.; Moroff, S.; Breen, M. Genomic profiling of canine mast cell tumors identifies DNA copy number aberrations associated with KIT mutations and high histological grade. Chromosome Res. 2017, 25, 129–143. [Google Scholar] [CrossRef]
- Tanvetthayanont, P.; Yata, T.; Boonnil, J.; Temisak, S.; Ponglowhapan, S. Validation of droplet digital PCR for cytokeratin 19 mRNA detection in canine peripheral blood and mammary gland. Sci. Rep. 2022, 12, 13623. [Google Scholar] [CrossRef]
- Kehl, A.; Valkai, M.; Van de Weyer, A.L.; Brockmann, M.; Steiger, K.; Schusser, B.; Aupperle-Lellbach, H. miRNA Profiles of Canine Intestinal Carcinomas, Lymphomas and Enteritis Analysed by Digital Droplet PCR from FFPE Material. Vet. Sci. 2023, 10, 125. [Google Scholar] [CrossRef]
- Hussain, S.; Saxena, S.; Shrivastava, S.; Mohanty, A.K.; Kumar, S.; Singh, R.J.; Kumar, A.; Wani, S.A.; Gandham, R.K.; Kumar, N.; et al. Gene expression profiling of spontaneously occurring canine mammary tumours: Insight into gene networks and pathways linked to cancer pathogenesis. PLoS ONE 2018, 13, e0208656. [Google Scholar] [CrossRef]
- Selvarajah, G.T.; Kirpensteijn, J.; van Wolferen, M.E.; Rao, N.A.S.; Fieten, H.; Mol, J.A. Gene expression profiling of canine osteosarcoma reveals genes associated with short and long survival times. Molecular Cancer 2009, 8, 72. [Google Scholar] [CrossRef]
- Thomas, R.; Fiegler, H.; Ostrander, E.A.; Galibert, F.; Carter, N.P.; Breen, M. A canine cancer-gene microarray for CGH analysis of tumors. Cytogenet. Genome Res. 2003, 102, 254–260. [Google Scholar] [CrossRef]
- Brocca, G.; Ferraresso, S.; Zamboni, C.; Martinez-Merlo, E.M.; Ferro, S.; Goldschmidt, M.H.; Castagnaro, M. Array Comparative Genomic Hybridization Analysis Reveals Significantly Enriched Pathways in Canine Oral Melanoma. Front. Oncol. 2019, 9, 1397. [Google Scholar] [CrossRef] [PubMed]
- Laufer-Amorim, R.; Fonseca-Alves, C.E.; Villacis, R.A.R.; Linde, S.A.D.; Carvalho, M.; Larsen, S.J.; Marchi, F.A.; Rogatto, S.R. Comprehensive Genomic Profiling of Androgen-Receptor-Negative Canine Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1555. [Google Scholar] [CrossRef] [PubMed]
- Ferraresso, S.; Aricò, A.; Sanavia, T.; Da Ros, S.; Milan, M.; Cascione, L.; Comazzi, S.; Martini, V.; Giantin, M.; Di Camillo, B.; et al. DNA methylation profiling reveals common signatures of tumorigenesis and defines epigenetic prognostic subtypes of canine Diffuse Large B-cell Lymphoma. Sci. Rep. 2017, 7, 11591. [Google Scholar] [CrossRef]
- Roberts, R.A.; Sabalos, C.M.; LeBlanc, M.L.; Martel, R.R.; Frutiger, Y.M.; Unger, J.M.; Botros, I.W.; Rounseville, M.P.; Seligmann, B.E.; Miller, T.P.; et al. Quantitative nuclease protection assay in paraffin-embedded tissue replicates prognostic microarray gene expression in diffuse large-B-cell lymphoma. Lab. Investig. 2007, 87, 979–997. [Google Scholar] [CrossRef] [PubMed]
- Rimsza, L.M.; Leblanc, M.L.; Unger, J.M.; Miller, T.P.; Grogan, T.M.; Persky, D.O.; Martel, R.R.; Sabalos, C.M.; Seligmann, B.; Braziel, R.M.; et al. Gene expression predicts overall survival in paraffin-embedded tissues of diffuse large B-cell lymphoma treated with R-CHOP. Blood 2008, 112, 3425–3433. [Google Scholar] [CrossRef] [PubMed]
- Bourzac, K.M.; Rounseville, M.P.; Zarate, X.; Maddula, V.S.; Henderson, D.C.; Luckey, J.A.; Seligmann, B.; Galbraith, D.W. A high-density quantitative nuclease protection microarray platform for high throughput analysis of gene expression. J. Biotechnol. 2011, 154, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.; Schwartz, M.; Duchemin, D.; Carl Barrett, J.; Post, G. Validation of a Multiplexed Gene Signature Assay for Diagnosis of Canine Cancers from Formalin-Fixed Paraffin-Embedded Tissues. J. Vet. Intern. Med. 2017, 31, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Letard, S.; Yang, Y.; Hanssens, K.; Palmérini, F.; Leventhal, P.S.; Guéry, S.; Moussy, A.; Kinet, J.P.; Hermine, O.; Dubreuil, P. Gain-of-function mutations in the extracellular domain of KIT are common in canine mast cell tumors. Mol. Cancer Res. 2008, 6, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.; Kehl, A.; Müller, T.; Klopfleisch, R.; Aupperle-Lellbach, H. Immunohistochemical and Molecular Genetic Analysis of Canine Digital Mast Cell Tumours. Animals 2023, 13, 1694. [Google Scholar] [CrossRef] [PubMed]
- Peralta, S.; Webb, S.M.; Katt, W.P.; Grenier, J.K.; Duhamel, G.E. Highly recurrent BRAF p.V595E mutation in canine papillary oral squamous cell carcinoma. Vet. Comp. Oncol. 2023, 21, 138–144. [Google Scholar] [CrossRef]
- Kerick, M.; Isau, M.; Timmermann, B.; Sültmann, H.; Herwig, R.; Krobitsch, S.; Schaefer, G.; Verdorfer, I.; Bartsch, G.; Klocker, H.; et al. Targeted high throughput sequencing in clinical cancer settings: Formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med. Genomics 2011, 4, 68. [Google Scholar] [CrossRef]
- Dickinson, P.J.; York, D.; Higgins, R.J.; LeCouteur, R.A.; Joshi, N.; Bannasch, D. Chromosomal Aberrations in Canine Gliomas Define Candidate Genes and Common Pathways in Dogs and Humans. J. Neuropathol. Exp. Neurol. 2016, 75, 700–710. [Google Scholar] [CrossRef]
- Strakova, A.; Ní Leathlobhair, M.; Wang, G.D.; Yin, T.T.; Airikkala-Otter, I.; Allen, J.L.; Allum, K.M.; Bansse-Issa, L.; Bisson, J.L.; Castillo Domracheva, A.; et al. Mitochondrial genetic diversity, selection and recombination in a canine transmissible cancer. Elife 2016, 5, e14552. [Google Scholar] [CrossRef]
- Gardner, H.L.; Sivaprakasam, K.; Briones, N.; Zismann, V.; Perdigones, N.; Drenner, K.; Facista, S.; Richholt, R.; Liang, W.; Aldrich, J.; et al. Canine osteosarcoma genome sequencing identifies recurrent mutations in DMD and the histone methyltransferase gene SETD2. Commun. Biol. 2019, 2, 266. [Google Scholar] [CrossRef]
- Sparks, A.; Woods, J.P.; Bienzle, D.; Wood, G.A.; Coomber, B.L. Whole genome sequencing analysis of high confidence variants of B-cell lymphoma in Canis familiaris. PLoS ONE 2020, 15, e0238183. [Google Scholar] [CrossRef] [PubMed]
- Cronise, K.E.; Das, S.; Hernandez, B.G.; Regan, D.P.; Dailey, D.D.; McGeachan, R.I.; Lana, S.E.; Page, R.L.; Gustafson, D.L.; Duval, D.L. Characterizing the molecular and immune landscape of canine bladder cancer. Vet. Comp. Oncol. 2022, 20, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Vozdova, M.; Kubickova, S.; Pal, K.; Fröhlich, J.; Fictum, P.; Rubes, J. Recurrent gene mutations detected in canine mast cell tumours by next generation sequencing. Vet. Comp. Oncol. 2020, 18, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Yang, I.S.; Seung, B.J.; Lee, S.; Kim, D.; Ha, Y.J.; Seo, M.K.; Kim, K.K.; Kim, H.S.; Cheong, J.H.; et al. Cross-species oncogenic signatures of breast cancer in canine mammary tumors. Nat. Commun. 2020, 11, 3616. [Google Scholar] [CrossRef]
- Das, S.; Idate, R.; Lana, S.E.; Regan, D.P.; Duval, D.L. Integrated analysis of canine soft tissue sarcomas identifies recurrent mutations in TP53, KMT genes and PDGFB fusions. Sci. Rep. 2023, 13, 10422. [Google Scholar] [CrossRef]
- Megquier, K.; Turner-Maier, J.; Swofford, R.; Kim, J.H.; Sarver, A.L.; Wang, C.; Sakthikumar, S.; Johnson, J.; Koltookian, M.; Lewellen, M.; et al. Comparative Genomics Reveals Shared Mutational Landscape in Canine Hemangiosarcoma and Human Angiosarcoma. Mol. Cancer Res. 2019, 17, 2410–2421. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wu, M.; Durham, A.C.; Radaelli, E.; Mason, N.J.; Xu, X.; Roth, D.B. Molecular subtypes in canine hemangiosarcoma reveal similarities with human angiosarcoma. PLoS ONE 2020, 15, e0229728. [Google Scholar] [CrossRef]
- Wong, K.; Ludwig, L.; Krijgsman, O.; Adams, D.J.; Wood, G.A.; van der Weyden, L. Comparison of the oncogenomic landscape of canine and feline hemangiosarcoma shows novel parallels with human angiosarcoma. Dis. Model. Mech. 2021, 14, dmm049044. [Google Scholar] [CrossRef]
- Wong, S.; Ehrhart, E.J.; Stewart, S.; Zismann, V.; Cawley, J.; Halperin, R.; Briones, N.; Richter, K.; Sivaprakasam, K.; Perdigones, N.; et al. Genomic landscapes of canine splenic angiosarcoma (hemangiosarcoma) contain extensive heterogeneity within and between patients. PLoS ONE 2022, 17, e0264986. [Google Scholar] [CrossRef]
- Wang, G.; Wu, M.; Durham, A.C.; Mason, N.J.; Roth, D.B. Canine Oncopanel: A capture-based, NGS platform for evaluating the mutational landscape and detecting putative driver mutations in canine cancers. Vet. Comp. Oncol. 2022, 20, 91–101. [Google Scholar] [CrossRef]
- Ulvé, R.; Rault, M.; Bahin, M.; Lagoutte, L.; Abadie, J.; De Brito, C.; Coindre, J.M.; Botherel, N.; Rousseau, A.; Wucher, V.; et al. Discovery of Human-Similar Gene Fusions in Canine Cancers. Cancer Res. 2017, 77, 5721–5727. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Tani, A.; Goto-Koshino, Y.; Motegi, T.; Sakamoto, M.; Mochizuki, T.; Harada, K.; Kobayashi, T.; Setoguchi, A.; Shizuta, Y.; et al. Gene expression profiles associated with early relapse during first remission induction in canine multicentric high-grade B-cell lymphoma. J. Vet. Med. Sci. 2023, 86, 18–27. [Google Scholar] [CrossRef]
- Elshafie, N.O.; Gribskov, M.; Lichti, N.I.; Sayedahmed, E.E.; Childress, M.O.; Dos Santos, A.P. miRNome expression analysis in canine diffuse large B-cell lymphoma. Front. Oncol. 2023, 13, 1238613. [Google Scholar] [CrossRef]
- Barros-Silva, D.; Marques, C.J.; Henrique, R.; Jerónimo, C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes 2018, 9, 429. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Lee, K.H.; Nam, A.R.; Cho, J.Y. Genome-Wide Methylation Profiling in Canine Mammary Tumor Reveals miRNA Candidates Associated with Human Breast Cancer. Cancers 2019, 11, 1466. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.B.; Anderson, K.J.; Boudreau, C.E.; Martinez-Ledesma, E.; Kocakavuk, E.; Johnson, K.C.; Barthel, F.P.; Varn, F.S.; Kassab, C.; Ling, X.; et al. Comparative Molecular Life History of Spontaneous Canine and Human Gliomas. Cancer Cell 2020, 37, 243–257.e247. [Google Scholar] [CrossRef] [PubMed]
- Mills, L.J.; Scott, M.C.; Shah, P.; Cunanan, A.R.; Deshpande, A.; Auch, B.; Curtin, B.; Beckman, K.B.; Spector, L.G.; Sarver, A.L.; et al. Comparative analysis of genome-wide DNA methylation identifies patterns that associate with conserved transcriptional programs in osteosarcoma. Bone 2022, 158, 115716. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Nance, R.L.; Wang, X.; Sandey, M.; Matz, B.M.; Thomas, A.; Smith, B.F. Single-Nuclei Multiome (ATAC + Gene Expression) Sequencing of a Primary Canine Osteosarcoma Elucidates Intra-Tumoral Heterogeneity and Characterizes the Tumor Microenvironment. Int. J. Mol. Sci. 2023, 24, 16365. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Hoeppner, M.P.; Lundquist, A.; Pirun, M.; Meadows, J.R.; Zamani, N.; Johnson, J.; Sundström, G.; Cook, A.; FitzGerald, M.G.; Swofford, R.; et al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS ONE 2014, 9, e91172. [Google Scholar] [CrossRef]
- Kinsey, N.A.; Belanger, J.M.; Oberbauer, A.M. Canine reference genome accuracy impacts variant calling: Lessons learned from investigating embryonic lethal variants. Anim. Genet. 2022, 53, 706–708. [Google Scholar] [CrossRef]
- Wang, C.; Wallerman, O.; Arendt, M.L.; Sundström, E.; Karlsson, Å.; Nordin, J.; Mäkeläinen, S.; Pielberg, G.R.; Hanson, J.; Ohlsson, Å.; et al. A novel canine reference genome resolves genomic architecture and uncovers transcript complexity. Commun. Biol. 2021, 4, 185. [Google Scholar] [CrossRef]
- Jagannathan, V.; Hitte, C.; Kidd, J.M.; Masterson, P.; Murphy, T.D.; Emery, S.; Davis, B.; Buckley, R.M.; Liu, Y.H.; Zhang, X.Q.; et al. Dog10K_Boxer_Tasha_1.0: A Long-Read Assembly of the Dog Reference Genome. Genes 2021, 12, 847. [Google Scholar] [CrossRef]
- Liao, W.W.; Asri, M.; Ebler, J.; Doerr, D.; Haukness, M.; Hickey, G.; Lu, S.; Lucas, J.K.; Monlong, J.; Abel, H.J.; et al. A draft human pangenome reference. Nature 2023, 617, 312–324. [Google Scholar] [CrossRef]
- Parker, H.G.; Kim, L.V.; Sutter, N.B.; Carlson, S.; Lorentzen, T.D.; Malek, T.B.; Johnson, G.S.; DeFrance, H.B.; Ostrander, E.A.; Kruglyak, L. Genetic structure of the purebred domestic dog. Science 2004, 304, 1160–1164. [Google Scholar] [CrossRef]
- Ebert, P.; Audano, P.A.; Zhu, Q.; Rodriguez-Martin, B.; Porubsky, D.; Bonder, M.J.; Sulovari, A.; Ebler, J.; Zhou, W.; Serra Mari, R.; et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 2021, 372, eabf7117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Collins, R.L.; Lee, W.P.; Weber, A.M.; Jun, Y.; Zhu, Q.; Weisburd, B.; Huang, Y.; Audano, P.A.; Wang, H.; et al. Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. Am. J. Hum. Genet. 2021, 108, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Field, M.A.; Rosen, B.D.; Dudchenko, O.; Chan, E.K.F.; Minoche, A.E.; Edwards, R.J.; Barton, K.; Lyons, R.J.; Tuipulotu, D.E.; Hayes, V.M.; et al. Canfam_GSD: De novo chromosome-length genome assembly of the German Shepherd Dog (Canis lupus familiaris) using a combination of long reads, optical mapping, and Hi-C. Gigascience 2020, 9, giaa027. [Google Scholar] [CrossRef] [PubMed]
- Player, R.A.; Forsyth, E.R.; Verratti, K.J.; Mohr, D.W.; Scott, A.F.; Bradburne, C.E. A novel canis lupus familiaris reference genome improves variant resolution for use in breed-specific GWAS. Life Sci. Alliance 2021, 4, e202000902. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.J.; Field, M.A.; Ferguson, J.M.; Dudchenko, O.; Keilwagen, J.; Rosen, B.D.; Johnson, G.S.; Rice, E.S.; Hillier, D.; Hammond, J.M.; et al. Chromosome-length genome assembly and structural variations of the primal Basenji dog (Canis lupus familiaris) genome. BMC Genom. 2021, 22, 188. [Google Scholar] [CrossRef]
- Halo, J.V.; Pendleton, A.L.; Shen, F.; Doucet, A.J.; Derrien, T.; Hitte, C.; Kirby, L.E.; Myers, B.; Sliwerska, E.; Emery, S.; et al. Long-read assembly of a Great Dane genome highlights the contribution of GC-rich sequence and mobile elements to canine genomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2016274118. [Google Scholar] [CrossRef]
- Schall, P.Z.; Winkler, P.A.; Petersen-Jones, S.M.; Yuzbasiyan-Gurkan, V.; Kidd, J.M. Genome-wide methylation patterns from canine nanopore assemblies. G3 Genes Genomes Genet. 2023, 13, jkad203. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Min, J.L.; Huang, J.; Crooks, L.; Memari, Y.; McCarthy, S.; Perry, J.R.; Xu, C.; Futema, M.; Lawson, D.; et al. The UK10K project identifies rare variants in health and disease. Nature 2015, 526, 82–90. [Google Scholar] [CrossRef]
- Plassais, J.; Kim, J.; Davis, B.W.; Karyadi, D.M.; Hogan, A.N.; Harris, A.C.; Decker, B.; Parker, H.G.; Ostrander, E.A. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun. 2019, 10, 1489. [Google Scholar] [CrossRef] [PubMed]
- Meadows, J.R.S.; Kidd, J.M.; Wang, G.D.; Parker, H.G.; Schall, P.Z.; Bianchi, M.; Christmas, M.J.; Bougiouri, K.; Buckley, R.M.; Hitte, C.; et al. Genome sequencing of 2000 canids by the Dog10K consortium advances the understanding of demography, genome function and architecture. Genome Biol. 2023, 24, 187. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Knapp, D.W.; Dhawan, D.; Ramos-Vara, J.A.; Ratliff, T.L.; Cresswell, G.M.; Utturkar, S.; Sommer, B.C.; Fulkerson, C.M.; Hahn, N.M. Naturally-Occurring Invasive Urothelial Carcinoma in Dogs, a Unique Model to Drive Advances in Managing Muscle Invasive Bladder Cancer in Humans. Front. Oncol. 2019, 9, 1493. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.; Milner, R.J.; Cortés-Hinojosa, G.; Riva, A.; Bechtel, S.; Sahay, B.; Cascio, M.; Lejeune, A.; Shiomitsu, K.; Souza, C.; et al. Novel application of single-cell next-generation sequencing for determination of intratumoral heterogeneity of canine osteosarcoma cell lines. J. Vet. Diagn. Investig. 2021, 33, 261–278. [Google Scholar] [CrossRef]
- Ammons, D.T.; Harris, R.A.; Hopkins, L.S.; Kurihara, J.; Weishaar, K.; Dow, S. A single-cell RNA sequencing atlas of circulating leukocytes from healthy and osteosarcoma affected dogs. Front. Immunol. 2023, 14, 1162700. [Google Scholar] [CrossRef] [PubMed]
- Ammons, D.; Hopkins, L.; Cronise, K.; Kurihara, J.; Regan, D.; Dow, S. Single-cell RNA sequencing reveals the cellular and molecular heterogeneity of treatment-naïve primary osteosarcoma in dogs. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, A.J.; Houser, A.E.; Ji, A.L. Research Techniques Made Simple: Spatial Transcriptomics. J. Investig. Dermatol. 2022, 142, 993–1001.e1001. [Google Scholar] [CrossRef]
- Yu, Q.; Jiang, M.; Wu, L. Spatial transcriptomics technology in cancer research. Front. Oncol. 2022, 12, 1019111. [Google Scholar] [CrossRef]
- Hu, B.; Sajid, M.; Lv, R.; Liu, L.; Sun, C. A review of spatial profiling technologies for characterizing the tumor microenvironment in immuno-oncology. Front. Immunol. 2022, 13, 996721. [Google Scholar] [CrossRef]
- Arora, R.; Cao, C.; Kumar, M.; Sinha, S.; Chanda, A.; McNeil, R.; Samuel, D.; Arora, R.K.; Matthews, T.W.; Chandarana, S.; et al. Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response. Nat. Commun. 2023, 14, 5029. [Google Scholar] [CrossRef]
- De Biase, D.; Prisco, F.; Piegari, G.; Ilsami, A.; d’Aquino, I.; Baldassarre, V.; Zito Marino, F.; Franco, R.; Papparella, S.; Paciello, O. RNAScope in situ Hybridization as a Novel Technique for the Assessment of c-KIT mRNA Expression in Canine Mast Cell Tumor. Front. Vet. Sci. 2021, 8, 591961. [Google Scholar] [CrossRef]
- Church, S.; Sutton, B.; Piazza, E.; Teplitz, K. 6 Interrogation of spatial transcriptomics on canine tumor and normal tissue. J. ImmunoTherapy Cancer 2023, 11, A6. [Google Scholar] [CrossRef]
- Auersperg, N.; Pawson, T.; Worth, A.; Weinmaster, G. Modifications of tumor histology by point mutations in the v-fps oncogene: Possible role of extracellular matrix. Cancer Res. 1987, 47, 6341–6348. [Google Scholar]
- Saldanha, O.L.; Loeffler, C.M.L.; Niehues, J.M.; van Treeck, M.; Seraphin, T.P.; Hewitt, K.J.; Cifci, D.; Veldhuizen, G.P.; Ramesh, S.; Pearson, A.T.; et al. Self-supervised attention-based deep learning for pan-cancer mutation prediction from histopathology. NPJ Precis. Oncol. 2023, 7, 35. [Google Scholar] [CrossRef]
- Loeffler, C.M.L.; Gaisa, N.T.; Muti, H.S.; van Treeck, M.; Echle, A.; Ghaffari Laleh, N.; Trautwein, C.; Heij, L.R.; Grabsch, H.I.; Ortiz Bruechle, N.; et al. Predicting Mutational Status of Driver and Suppressor Genes Directly from Histopathology With Deep Learning: A Systematic Study Across 23 Solid Tumor Types. Front. Genet. 2021, 12, 806386. [Google Scholar] [CrossRef]
- Echle, A.; Rindtorff, N.T.; Brinker, T.J.; Luedde, T.; Pearson, A.T.; Kather, J.N. Deep learning in cancer pathology: A new generation of clinical biomarkers. Br. J. Cancer 2021, 124, 686–696. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, M.; Wang, S.; Li, X.; Sun, Y. Emerging role of deep learning-based artificial intelligence in tumor pathology. Cancer Commun. 2020, 40, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Foroughi Pour, A.; White, B.S.; Park, J.; Sheridan, T.B.; Chuang, J.H. Deep learning features encode interpretable morphologies within histological images. Sci. Rep. 2022, 12, 9428. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Nomikou, S.; Coudray, N.; Jour, G.; Dawood, Z.; Hong, R.; Esteva, E.; Sakellaropoulos, T.; Donnelly, D.; Moran, U.; et al. Deep Learning and Pathomics Analyses Reveal Cell Nuclei as Important Features for Mutation Prediction of BRAF-Mutated Melanomas. J. Investig. Dermatol. 2022, 142, 1650–1658.e1656. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.; Raza, S.E.A.; Azam, A.; Graham, S.; Ilyas, M.; Cree, I.A.; Snead, D.; Minhas, F.; Rajpoot, N.M. Development and validation of a weakly supervised deep learning framework to predict the status of molecular pathways and key mutations in colorectal cancer from routine histology images: A retrospective study. Lancet Digit. Health 2021, 3, e763–e772. [Google Scholar] [CrossRef] [PubMed]
- Saltz, J.; Gupta, R.; Hou, L.; Kurc, T.; Singh, P.; Nguyen, V.; Samaras, D.; Shroyer, K.R.; Zhao, T.; Batiste, R.; et al. Spatial Organization and Molecular Correlation of Tumor-Infiltrating Lymphocytes Using Deep Learning on Pathology Images. Cell Rep. 2018, 23, 181–193.e187. [Google Scholar] [CrossRef]
- Tsou, P.; Wu, C.J. Mapping Driver Mutations to Histopathological Subtypes in Papillary Thyroid Carcinoma: Applying a Deep Convolutional Neural Network. J. Clin. Med. 2019, 8, 1675. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef]
- Fu, Y.; Jung, A.W.; Torne, R.V.; Gonzalez, S.; Vöhringer, H.; Shmatko, A.; Yates, L.R.; Jimenez-Linan, M.; Moore, L.; Gerstung, M. Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nat. Cancer 2020, 1, 800–810. [Google Scholar] [CrossRef]
- Wilm, F.; Fragoso, M.; Marzahl, C.; Qiu, J.; Puget, C.; Diehl, L.; Bertram, C.A.; Klopfleisch, R.; Maier, A.; Breininger, K.; et al. Pan-tumor CAnine cuTaneous Cancer Histology (CATCH) dataset. Sci. Data 2022, 9, 588. [Google Scholar] [CrossRef]
- Fragoso-Garcia, M.; Wilm, F.; Bertram, C.A.; Merz, S.; Schmidt, A.; Donovan, T.; Fuchs-Baumgartinger, A.; Bartel, A.; Marzahl, C.; Diehl, L.; et al. Automated diagnosis of 7 canine skin tumors using machine learning on H&E-stained whole slide images. Vet. Pathol. 2023, 60, 865–875. [Google Scholar] [CrossRef]
- Patkar, S.; Beck, J.; Harmon, S.; Mazcko, C.; Turkbey, B.; Choyke, P.; Brown, G.T.; LeBlanc, A. Deep Domain Adversarial Learning for Species-Agnostic Classification of Histologic Subtypes of Osteosarcoma. Am. J. Pathol. 2023, 193, 60–72. [Google Scholar] [CrossRef]
- Küchler, L.; Posthaus, C.; Jäger, K.; Guscetti, F.; van der Weyden, L.; von Bomhard, W.; Schmidt, J.M.; Farra, D.; Aupperle-Lellbach, H.; Kehl, A.; et al. Artificial Intelligence to Predict the BRAF V595E Mutation in Canine Urinary Bladder Urothelial Carcinomas. Animals 2023, 13, 2404. [Google Scholar] [CrossRef] [PubMed]
- Shmatko, A.; Ghaffari Laleh, N.; Gerstung, M.; Kather, J.N. Artificial intelligence in histopathology: Enhancing cancer research and clinical oncology. Nat. Cancer 2022, 3, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Rösler, W.; Altenbuchinger, M.; Baeßler, B.; Beissbarth, T.; Beutel, G.; Bock, R.; von Bubnoff, N.; Eckardt, J.N.; Foersch, S.; Loeffler, C.M.L.; et al. An overview and a roadmap for artificial intelligence in hematology and oncology. J. Cancer Res. Clin. Oncol. 2023, 149, 7997–8006. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.; Wu, E.; Zhang, A.; Alizadeh, A.A.; Zou, J. From patterns to patients: Advances in clinical machine learning for cancer diagnosis, prognosis, and treatment. Cell 2023, 186, 1772–1791. [Google Scholar] [CrossRef] [PubMed]
- Abele, N.; Tiemann, K.; Krech, T.; Wellmann, A.; Schaaf, C.; Länger, F.; Peters, A.; Donner, A.; Keil, F.; Daifalla, K.; et al. Noninferiority of Artificial Intelligence-Assisted Analysis of Ki-67 and Estrogen/Progesterone Receptor in Breast Cancer Routine Diagnostics. Mod. Pathol. 2023, 36, 100033. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, L.; Lv, L.; Fu, C.C.; Jin, Y.; Zheng, Q.; Wang, B.; Ye, Q.; Fang, Q.; Li, Y. A new AI-assisted scoring system for PD-L1 expression in NSCLC. Comput. Methods Programs Biomed. 2022, 221, 106829. [Google Scholar] [CrossRef] [PubMed]
- Sandøe, P.; Palmer, C.; Corr, S.A.; Springer, S.; Lund, T.B. Do people really care less about their cats than about their dogs? A comparative study in three European countries. Front. Vet. Sci. 2023, 10, 1237547. [Google Scholar] [CrossRef] [PubMed]



| Platform | Sequencing Technology | Considerations |
|---|---|---|
| Illumina | Uses a sequencing-by-synthesis approach whereby DNA fragments immobilised on a flow cell are amplified into clonal clusters, and the fluorescent signal released during the incorporation of the fluorescently tagged nucleotides into the growing DNA strand is translated into a base call |
|
| Ion Torrent | Uses a sequencing-by-synthesis approach whereby hydrogen ions released during the incorporation of a nucleotide into the growing DNA strand are detected and translated into a base call |
|
| Oxford Nanopore Technologies | Uses nanopore sequencing, whereby the change in electrical current made by a single DNA molecule passing through a nanopore is detected and translated into a base call |
|
| Pacific Biosciences (‘PacBio’) | Uses single-molecule real-time (SMRT) sequencing, which allows for the sequencing of individual DNA molecules in real time |
|
| Method | Advantages | Disadvantages |
|---|---|---|
| WGS |
|
|
| WES |
|
|
| TGS |
|
|
| Molecular Technology | Method | Use | Comments | |
|---|---|---|---|---|
| PCR | PCR (DNA), RT-PCR (RNA) | Amplifying specific regions of DNA/RNA | Amplification of region of interest for further analyses, such as quantification or sequencing | Fast and cheap |
| qPCR (DNA), RT-qPCR (RNA) | Real-time detection and quantification of specific DNA/RNA regions by fluorescence intensity | qPCR: detection of SNVs or small indels RT-qPCR: gene expression profiling | Fast and cheap | |
| dPCR (DNA, RNA) | Absolute quantification of specific DNA/RNA regions by fluorescence signal in droplets | dPCR: mutation analysis dPCR with RT-PCR: mRNA and miRNA expression quantification | Higher sensitivity than qPCR | |
| DNA microarray | Gene expression | Nucleic acid fragments labelled with a fluorescence dye by PCR or RT-PCR on a solid surface | Measurement of thousands of RNA transcripts in a single experiment | High throughput |
| aCGH | Quantitatively compares the fluorescence signal intensity from test DNA and control DNA | Detection of CNVs | Cheaper than NGS | |
| Nuclease protection assay | qNPA | After hybridisation with a probe, the targeted transcript is transferred to an array plate | Detection and quantification of mRNA expression | Highly suited for FFPE samples |
| Sanger sequencing | PCR including fluorophore-labelled nucleotides and capillary gel electrophoresis of products | Determines the DNA sequence of individual exons or genes | Fast, cheap | |
| NGS (DNA) | WGS | Creating a library of the sample by PCR with molecular barcodes, selection of regions of interest for WES or TGS by RNA capture probes ‘baits’, then clonal amplification by PCR, library sequencing and data analysis | Sequencing all nucleotides of the genome including chromosomal and mitochondrial DNA | See Table 2 |
| WES | Sequencing the entire coding region (i.e., all the exons) | |||
| TGS | Sequencing a selected portion of the genome (i.e., genes of interest) | |||
| NGS (RNA) | RNA-Seq | As NGS for DNA, but with an additional step for creating cDNA | Gene expression profiles, alternative splicing events, allele-specific expression and gene fusions | High sensitivity and reproducibility |
| Methylation sequencing | RBBS, ATAC-Seq, Microarray-based methylation profiling | Different techniques to determine the methylation of cytosines and ‘open’ regions of chromatin | Analysing the methylation profile and chromatin accessibility | For epigenetic investigations |
| Spatial transcriptomics | ISH-, ISS- and ISC-based methods | In situ mRNA investigation by FISH or sequencing | Transcriptomics in situ | Investigating the cell origin of an mRNA profile |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kehl, A.; Aupperle-Lellbach, H.; de Brot, S.; van der Weyden, L. Review of Molecular Technologies for Investigating Canine Cancer. Animals 2024, 14, 769. https://doi.org/10.3390/ani14050769
Kehl A, Aupperle-Lellbach H, de Brot S, van der Weyden L. Review of Molecular Technologies for Investigating Canine Cancer. Animals. 2024; 14(5):769. https://doi.org/10.3390/ani14050769
Chicago/Turabian StyleKehl, Alexandra, Heike Aupperle-Lellbach, Simone de Brot, and Louise van der Weyden. 2024. "Review of Molecular Technologies for Investigating Canine Cancer" Animals 14, no. 5: 769. https://doi.org/10.3390/ani14050769
APA StyleKehl, A., Aupperle-Lellbach, H., de Brot, S., & van der Weyden, L. (2024). Review of Molecular Technologies for Investigating Canine Cancer. Animals, 14(5), 769. https://doi.org/10.3390/ani14050769