
Comparison of Nucleosome Landscapes Between Porcine Embryonic Fibroblasts and GV Oocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Generation
2.2. Calculation of Global Nucleosome Occupancy
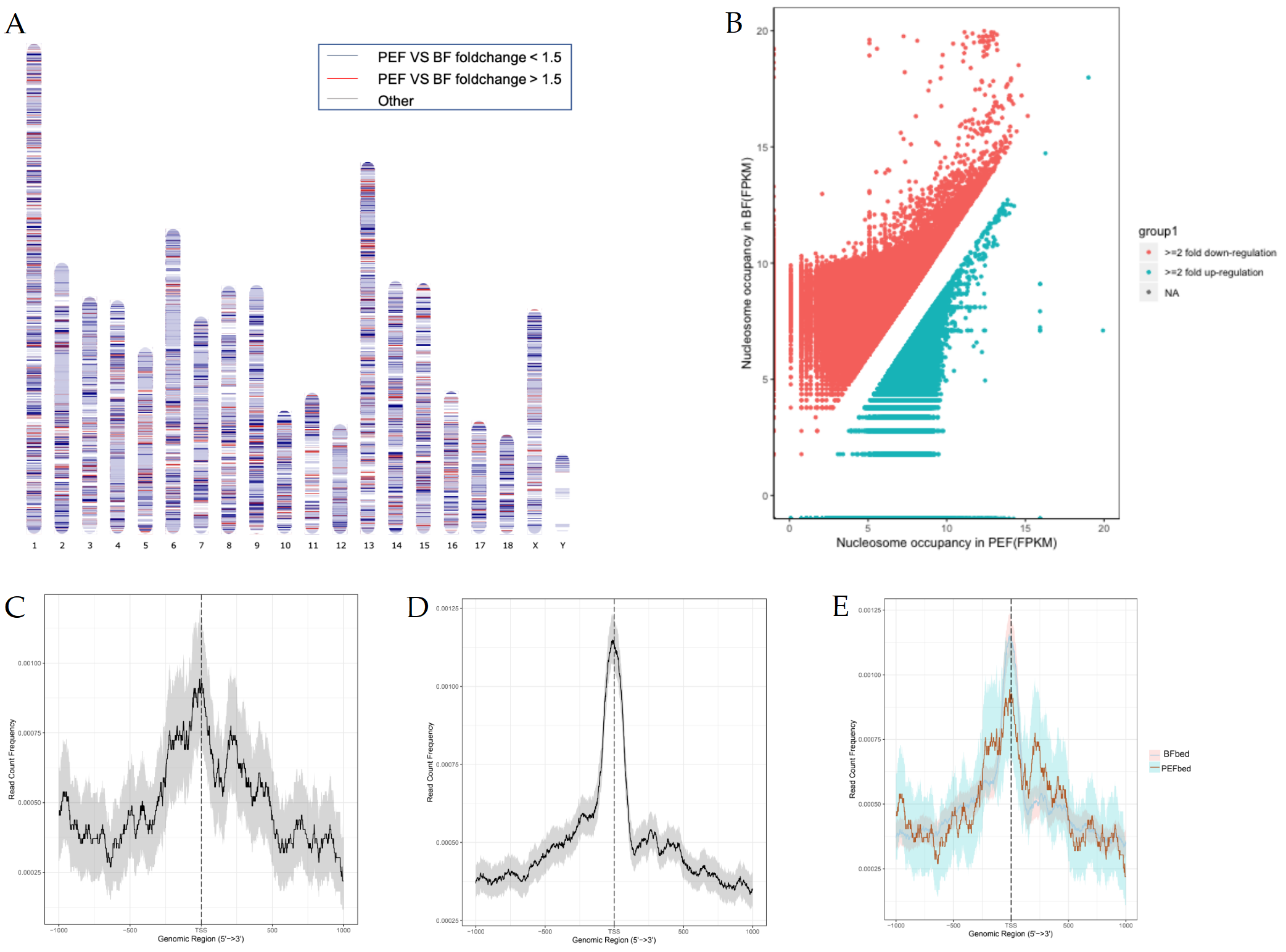
2.3. Genome-Wide Comparison of Nucleosome Occupancy
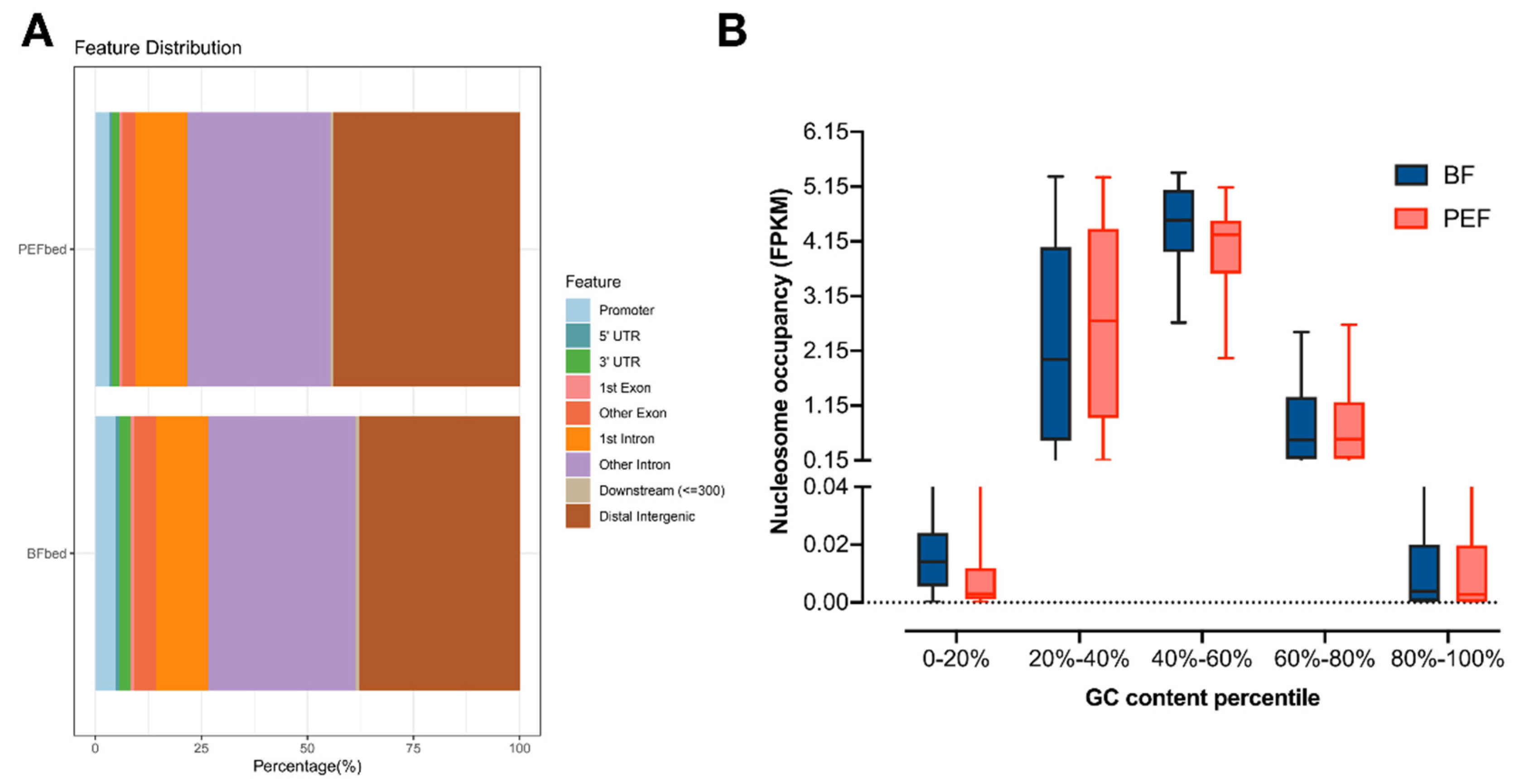
2.4. Nucleosome Occupancy in Different Regions of the Genome
2.5. Nucleosome Distribution Profiles
2.6. Analysis of Gene Expression
2.7. Statistical Analysis
3. Results
3.1. Global Nucleosome Organization and Occupancy of Fully Grown GV Oocytes and PEF
3.2. Different Nucleosome Occupancy Between the Two Cell Types
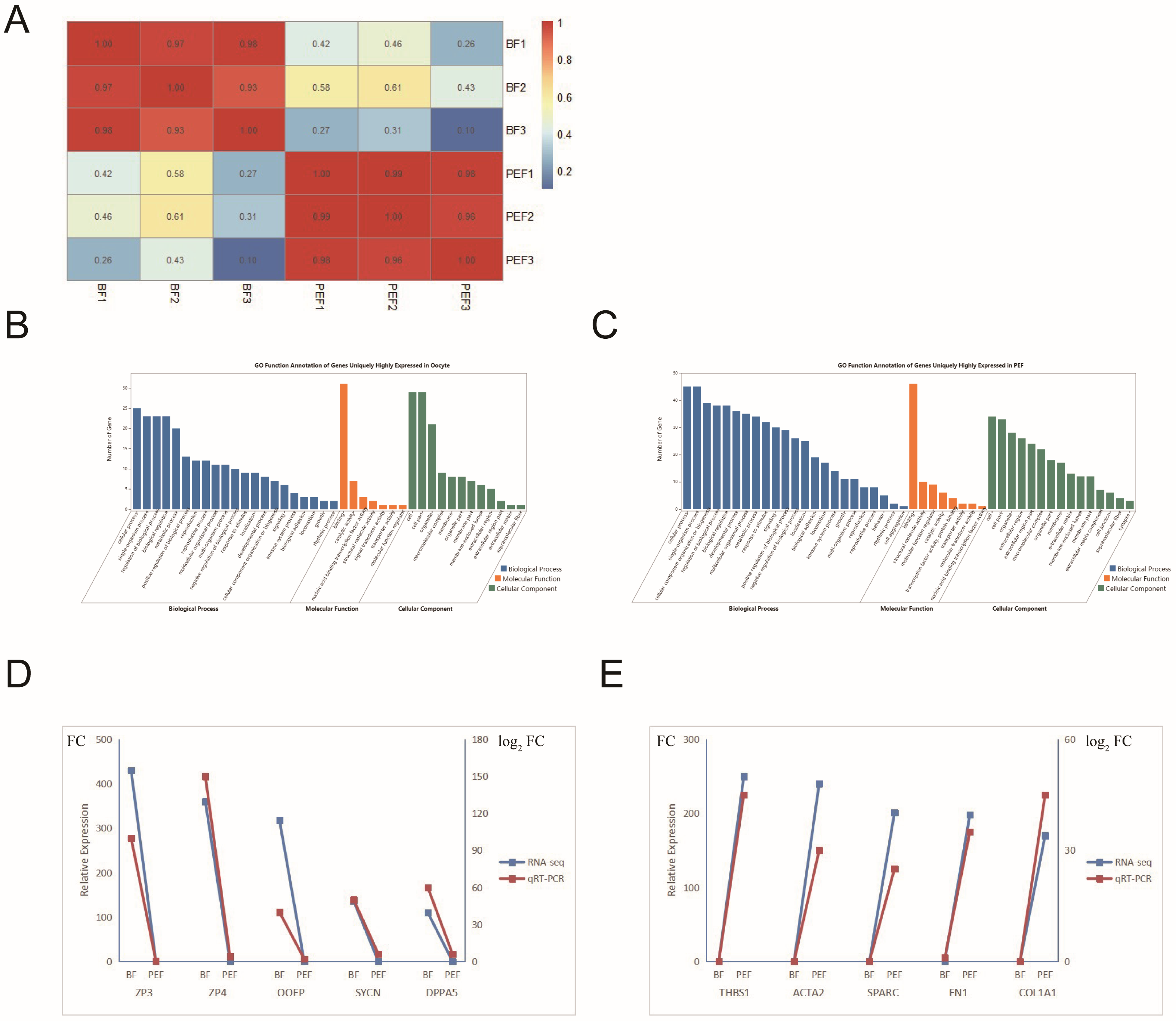
3.3. Specific Gene Expression in Two Cell Types
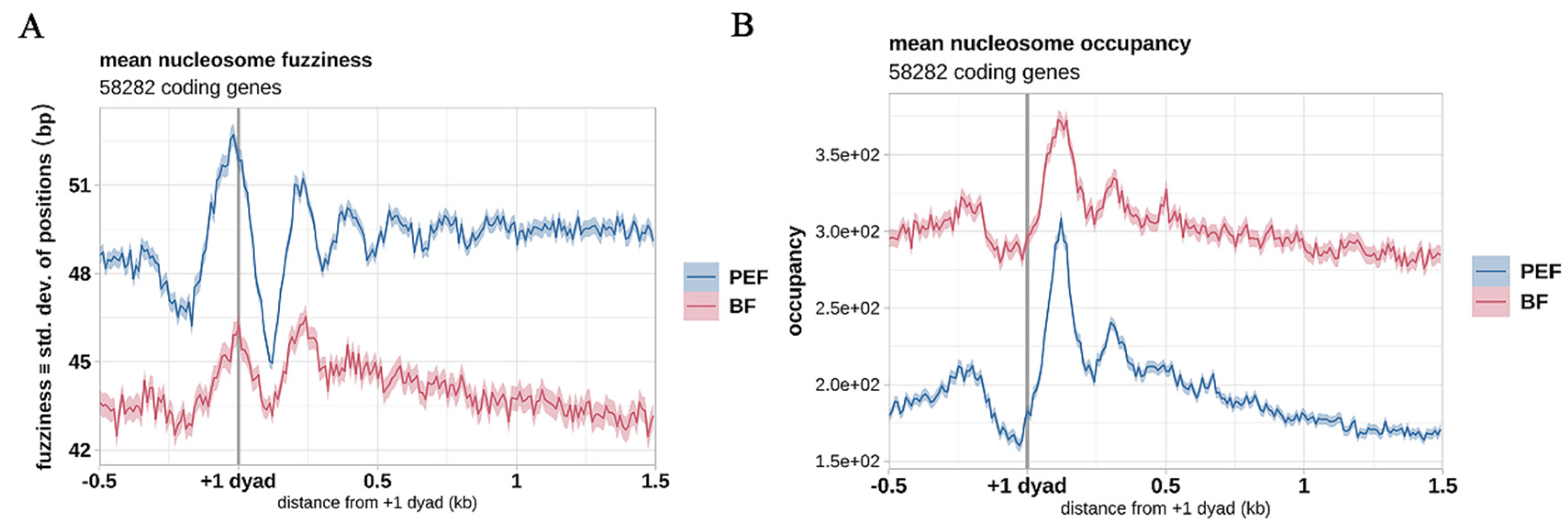
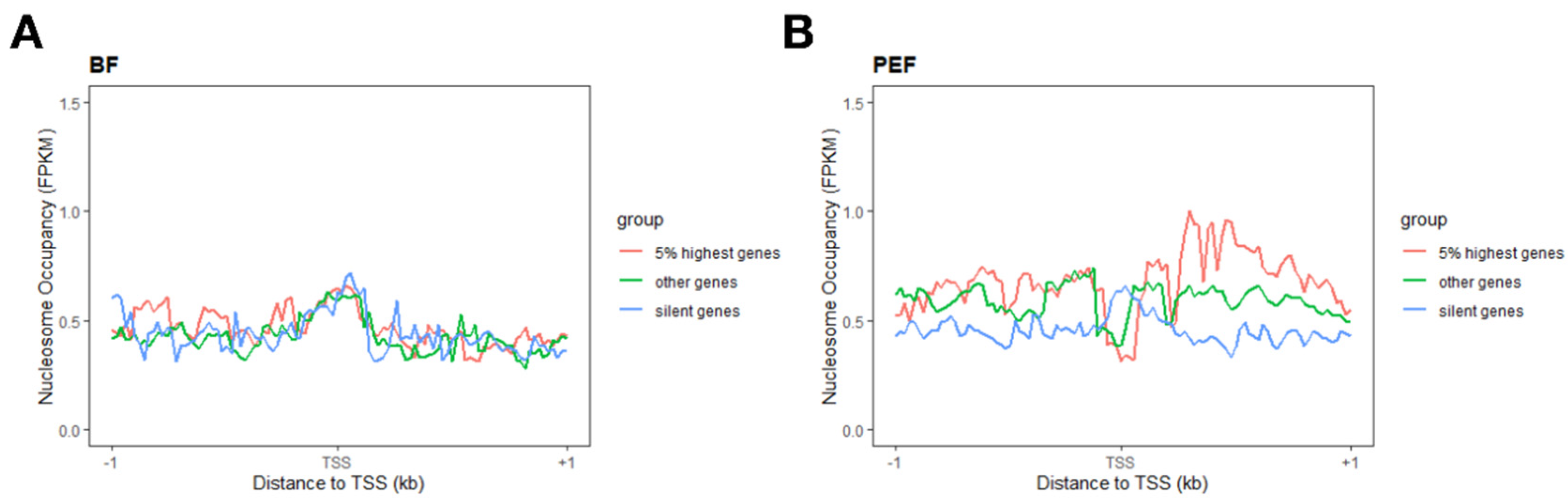
3.4. Nucleosome Configurations Around TSSs Correlate with Gene Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Field, Y.; Kaplan, N.; Fondufe-Mittendorf, Y.; Moore, I.K.; Sharon, E.; Lubling, Y.; Widom, J.; Segal, E. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput. Biol. 2008, 4, e1000216. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.; Moore, I.K.; Fondufe-Mittendorf, Y.; Gossett, A.J.; Tillo, D.; Field, Y.; LeProust, E.M.; Hughes, T.R.; Lieb, J.D.; Widom, J.; et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 2009, 458, 362–366. [Google Scholar] [CrossRef]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thåström, A.; Field, Y.; Moore, I.K.; Wang, J.-P.Z.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef]
- Lee, W.; Tillo, D.; Bray, N.; Morse, R.H.; Davis, R.W.; Hughes, T.R.; Nislow, C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet. 2007, 39, 1235–1244. [Google Scholar] [CrossRef]
- Tao, C.; Li, J.; Chen, B.; Chi, D.; Zeng, Y.; Liu, H. Genome-scale identification of nucleosome organization by using 1000 porcine oocytes at different developmental stages. PLoS ONE 2017, 12, e0174225. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Meldrum, D.R.; Katz-Jaffe, M.G.; Krisher, R.L.; Schoolcraft, W.B. Oocyte environment: Follicular fluid and cumulus cells are critical for oocyte health. Fertil. Steril. 2015, 103, 303–316. [Google Scholar] [CrossRef]
- Maekawa, M.; Yamaguchi, K.; Nakamura, T.; Shibukawa, R.; Kodanaka, I.; Ichisaka, T.; Kawamura, Y.; Mochizuki, H.; Goshima, N.; Yamanaka, S. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 2011, 474, 225–229. [Google Scholar] [CrossRef]
- Takahashi, K.; Sakurai, N.; Emura, N.; Hashizume, T.; Sawai, K. Effects of downregulating GLIS1 transcript on preimplantation development and gene expression of bovine embryos. J. Reprod. Dev. 2015, 61, 369–374. [Google Scholar] [CrossRef]
- Rajkovic, A.; Yan, C.; Yan, W.; Klysik, M.; Matzuk, M.M. Obox, a family of homeobox genes preferentially expressed in germ cells. Genomics 2002, 79, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-M.; Ahn, H.-J.; Lee, H.-S.; Lee, K.-A.; Lee, S.-M.; Kim, H.-H.; Kim, K.-S.; Park, K.-S. Obox4 regulates the expression of histone family genes and promotes differentiation of mouse embryonic stem cells. FEBS Lett. 2010, 584, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Hsieh-Li, H.M.; Yeh, Y.; Li, H. Mice lacking the Obox6 homeobox gene undergo normal early embryonic development and are fertile. Dev. Dyn. 2007, 236, 2636–2642. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.R.; Pálfy, M.; Hilbert, L.; Kumar, M.; Karschau, J.; Zaburdaev, V.; Shevchenko, A.; Vastenhouw, N.L. Competition between histone and transcription factor binding regulates the onset of transcription in zebrafish embryos. Elife 2017, 6, e23326. [Google Scholar] [CrossRef] [PubMed]
- Almouzni, G.; Wolffe, A. Constraints on transcriptional activator function contribute to transcriptional quiescence during early Xenopus embryogenesis. EMBO J. 1995, 14, 1752–1765. [Google Scholar] [CrossRef]
- Tao, Y.; Zheng, W.; Jiang, Y.; Ding, G.; Hou, X.; Tang, Y.; Li, Y.; Gao, S.; Chang, G.; Zhang, X.; et al. Nucleosome organizations in induced pluripotent stem cells reprogrammed from somatic cells belonging to three different germ layers. BMC Biol. 2014, 12, 109. [Google Scholar] [CrossRef]
- Bebbere, D.; Albertini, D.F.; Coticchio, G.; Borini, A.; Ledda, S. The subcortical maternal complex: Emerging roles and novel per-spectives. Mol. Hum. Reprod. 2021, 27, gaab043. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, X.; Shi, J.; Yao, M.; Lin, J.; Li, J.; Liu, H.; Li, H.; Shi, G.; Wang, Z.; et al. Dynamically reorganized chromatin is the key for the reprogramming of somatic cells to plu-ripotent cells. Sci. Rep. 2015, 5, 17691. [Google Scholar] [CrossRef]
- Qian, X.; Kim, J.K.; Tong, W.; Villa-Diaz, L.G.; Krebsbach, P.H. DPPA5 Supports Pluripotency and Reprogramming by Regulating NANOG Turnover. Stem Cells 2016, 34, 588–600. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, H.; Jiang, H.; Zhang, M.; Wang, J.; Zhao, L.; Wang, C.; Liu, M.; Li, R. Conversion between porcine naive-like and primed ESCs and specific pluripotency marker identification. Vitr. Cell. Dev. Biol.-Anim. 2020, 56, 412–423. [Google Scholar] [CrossRef]
- De Iaco, A.; Verp, S.; Offner, S.; Grun, D.; Trono, D. DUX is a non-essential synchronizer of zygotic genome activation. Development 2020, 147, dev177725. [Google Scholar] [CrossRef]
- Miharada, K.; Sigurdsson, V.; Karlsson, S. Dppa5 improves hematopoietic stem cell activity by reducing endoplasmic reticulum stress. Cell Rep. 2014, 7, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Jullien, J.; Pasque, V.; Halley-Stott, R.P.; Miyamoto, K.; Gurdon, J.B. Mechanisms of nuclear reprogramming by eggs and oocytes: A deterministic process? Nat. Rev. Mol. Cell. Biol. 2011, 12, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.; Li, J.; Zhang, X.; Chen, B.; Chi, D.; Zeng, Y.; Niu, Y.; Wang, C.; Cheng, W.; Wu, W.; et al. Dynamic Reorganization of Nucleosome Positioning in Somatic Cells after Transfer into Porcine Enucleated Oocytes. Stem Cell Rep. 2017, 9, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.R.; Wengier, D.L.; Bergmann, D.C. Cell-type-specific transcriptome and histone modification dynamics during cellular re-programming in the Arabidopsis stomatal lineage. Proc. Natl. Acad. Sci. USA 2019, 116, 21914–21924. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, H.; Pugh, B.F. Stable and dynamic nucleosome states during a meiotic developmental process. Genome Res. 2011, 21, 875–884. [Google Scholar] [CrossRef]
- Bouniol-Baly, C.; Hamraoui, L.; Guibert, J.; Beaujean, N.; Szöllösi, M.S.; Debey, P. Differential transcriptional activity associated with chromatin configuration in fully grown mouse germinal vesicle oocytes. Biol. Reprod. 1999, 60, 580–587. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Zhao, S.; Pang, Z.; Jia, C.; Tao, C. Comparison of Nucleosome Landscapes Between Porcine Embryonic Fibroblasts and GV Oocytes. Animals 2024, 14, 3392. https://doi.org/10.3390/ani14233392
Zhao M, Zhao S, Pang Z, Jia C, Tao C. Comparison of Nucleosome Landscapes Between Porcine Embryonic Fibroblasts and GV Oocytes. Animals. 2024; 14(23):3392. https://doi.org/10.3390/ani14233392
Chicago/Turabian StyleZhao, Minjun, Shunran Zhao, Zhaoqi Pang, Chunhui Jia, and Chenyu Tao. 2024. "Comparison of Nucleosome Landscapes Between Porcine Embryonic Fibroblasts and GV Oocytes" Animals 14, no. 23: 3392. https://doi.org/10.3390/ani14233392
APA StyleZhao, M., Zhao, S., Pang, Z., Jia, C., & Tao, C. (2024). Comparison of Nucleosome Landscapes Between Porcine Embryonic Fibroblasts and GV Oocytes. Animals, 14(23), 3392. https://doi.org/10.3390/ani14233392