
Genome-Wide Association Studies of Growth Trait Heterosis in Crossbred Meat Rabbits

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Genotypes
2.2. ROH Calling and Inbreeding Coefficients
2.3. Estimation of Individual Heterosis at Different Growth Time Points
2.4. Heterosis Evaluation of Growth Traits
3. Results
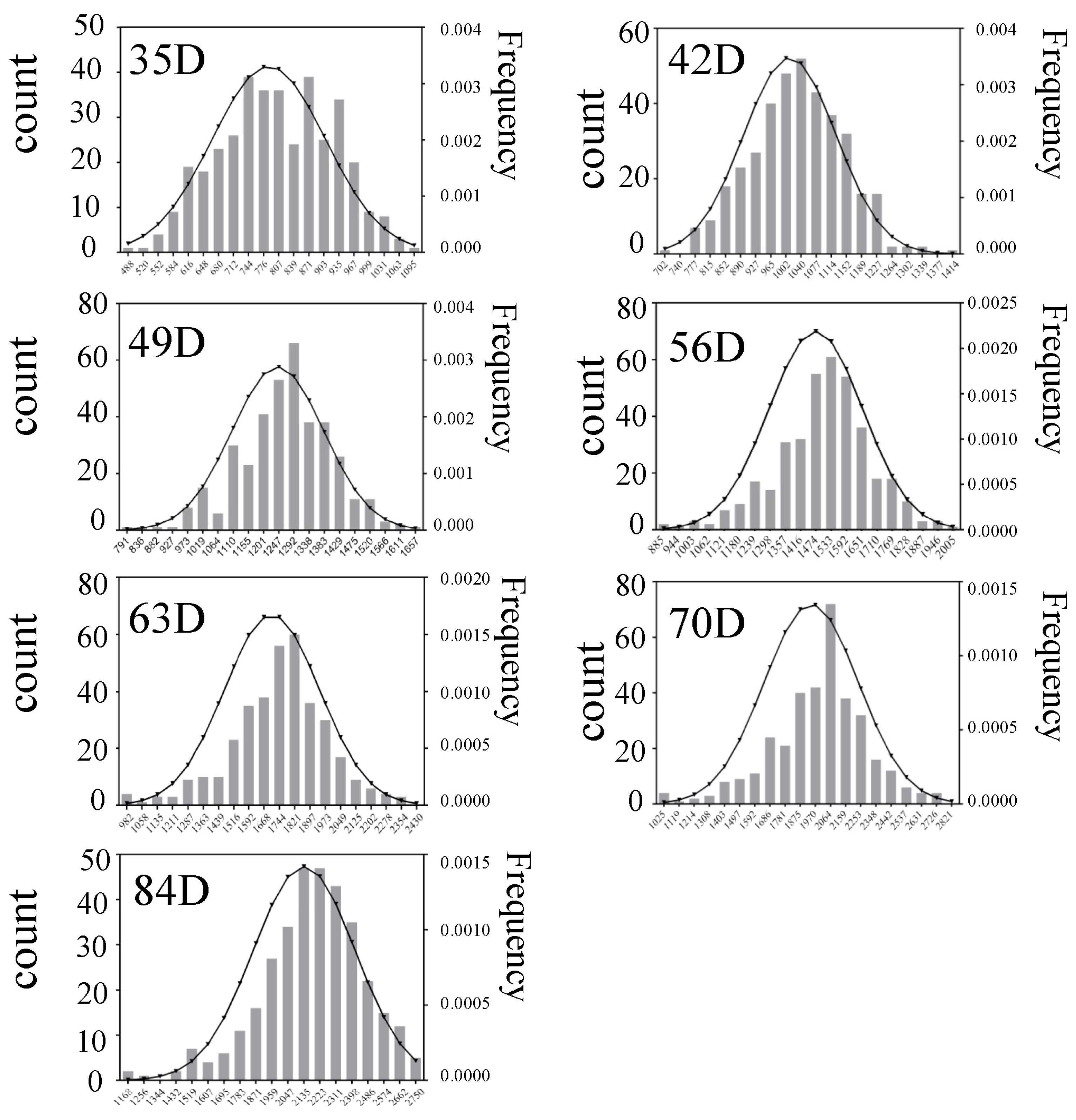
3.1. Phenotypic Data Analysis
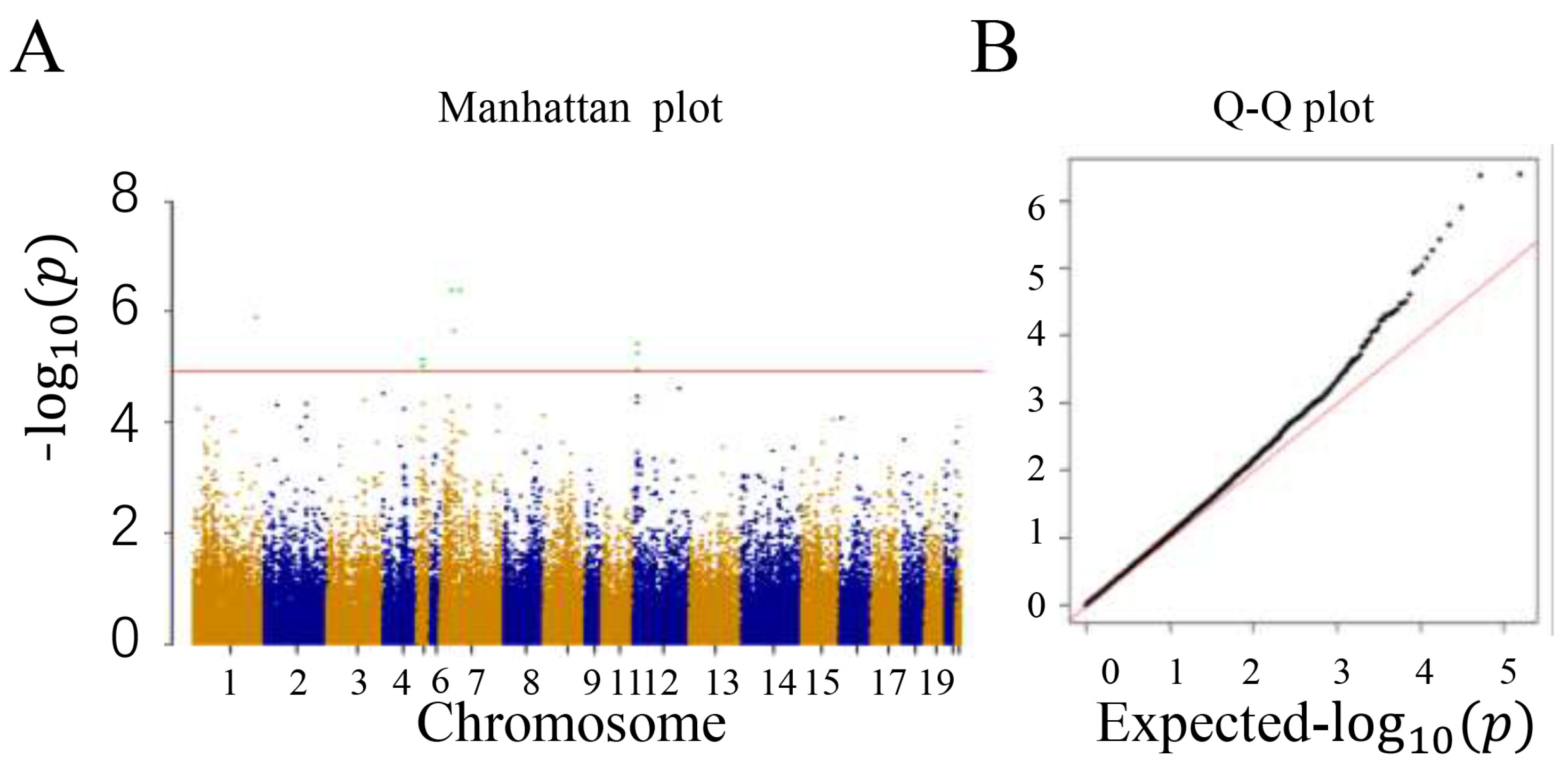
3.2. Genotyping and Population Stratification
3.3. Correlation Analysis of Growth Traits
3.4. ROH and Correlation of Inbreeding Coefficients
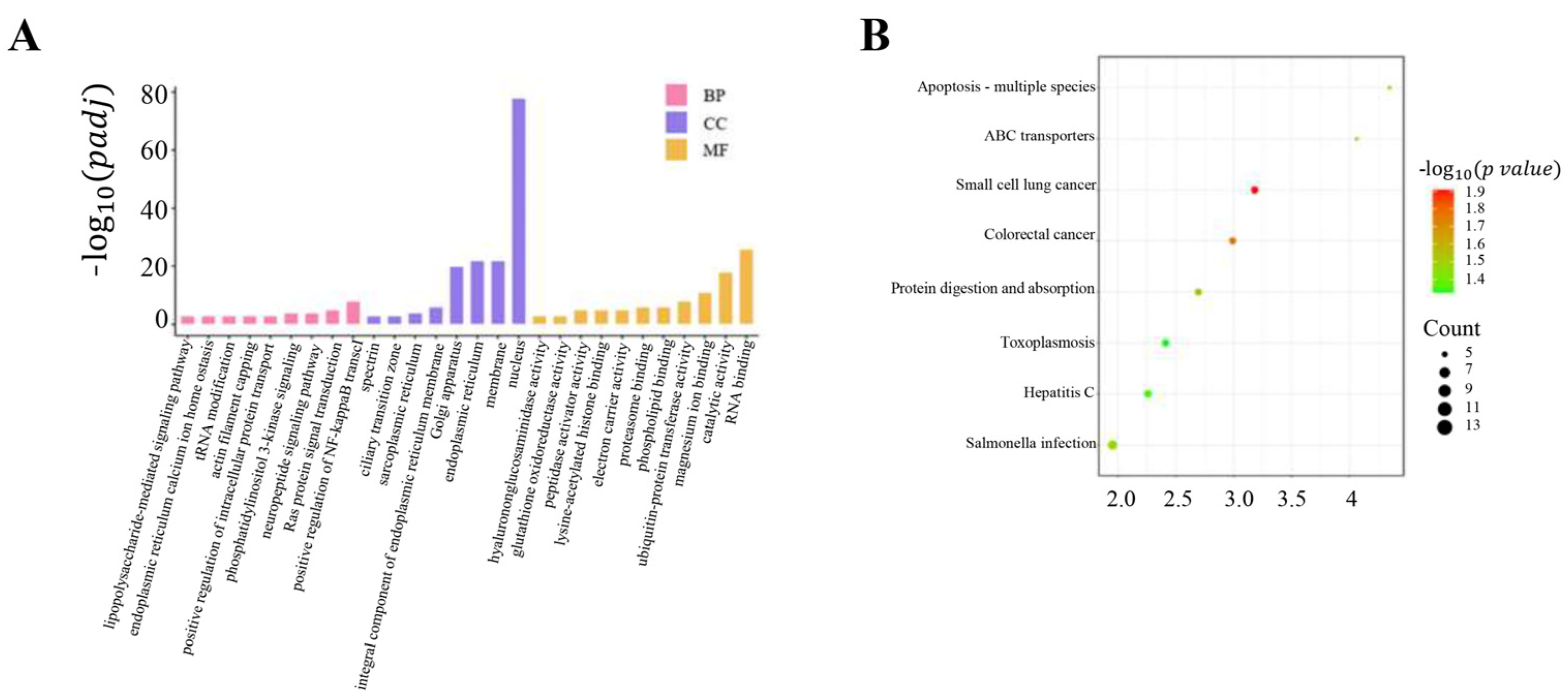
3.5. Functional Annotation of Candidate Genes in ROHs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cullere, M.; Dalle Zotte, A. Rabbit meat production and consumption: State of knowledge and future perspectives. Meat Sci. 2018, 143, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Nasr, M.A.F.; Abd-Elhamid, T.; Hussein, M.A. Growth performance, carcass characteristics, meat quality and muscle amino-acid profile of different rabbits breeds and their crosses. Meat Sci. 2017, 134, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, K.D.; Oladele, T.O.; Atolani, O.; Sulaimon, R.O.; Zubair, J.I.; Ajao, B.H. Dietary Plukenetia conophora seed alters muscle cholesterol, antioxidant enzymes, and fatty acids in growing rabbits. Meat Sci. 2022, 189, 108809. [Google Scholar] [CrossRef] [PubMed]
- Dalle Zotte, A.; Szendro, Z. The role of rabbit meat as functional food. Meat Sci. 2011, 88, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Risch, N.; Merikangas, K. The future of genetic studies of complex human diseases. Science 1996, 273, 1516–1517. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.-Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Barazandeh, A.; Mohammadabadi, M.R.; Ghaderi-Zefrehei, M.; Nezamabadi-pour, H. Genome-wide analysis of CpG islands in some livestock genomes and their relationship with genomic features. Czech J. Anim. Sci. 2016, 61, 487–495. [Google Scholar] [CrossRef]
- Bordbar, F.; Mohammadabadi, M.; Jensen, J.; Xu, L.; Li, J.; Zhang, L. Identification of Candidate Genes Regulating Carcass Depth and Hind Leg Circumference in Simmental Beef Cattle Using Illumina Bovine Beadchip and Next-Generation Sequencing Analyses. Animals 2022, 12, 1103. [Google Scholar] [CrossRef] [PubMed]
- Safaei, S.M.H.; Dadpasand, M.; Mohammadabadi, M.; Atashi, H.; Stavetska, R.; Klopenko, N.; Kalashnyk, O. An Origanum majora-na Leaf Diet Influences Myogenin Gene Expression, Performance, and Carcass Characteristics in Lambs. Animals 2022, 13, 14. [Google Scholar] [CrossRef]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef]
- Kim, E.-S.; Sonstegard, T.S.; Van Tassell, C.P.; Wiggans, G.; Rothschild, M.F. The Relationship between Runs of Homozygosity and Inbreeding in Jersey Cattle under Selection. PLoS ONE 2015, 10, e0129967. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Ciani, E.; Sardina, M.T.; Sottile, G.; Pilla, F.; Portolano, B.; Bi, O. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim. Genet. 2018, 49, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of Artificial Selection on Runs of Homozygosity in, U.S. Holstein Cattle. PLoS ONE 2013, 8, e80813. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic Patterns of Homozygosity in Worldwide Human Populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Kardos, M.; Qvarnström, A.; Ellegren, H. Inferring Individual Inbreeding and Demographic History from Segments of Identity by Descent in Ficedula Flycatcher Genome Sequences. Genetics 2017, 205, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Adams, S.M.; Derks, M.F.L.; Makanjuola, B.O.; Marras, G.; Wood, B.J.; Baes, C.F. Investigating inbreeding in the turkey (Meleagris gallopavo) genome. Poult. Sci. 2021, 100, 101366. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Coefficients of Inbreeding and Relationship. Am. Nat. 1922, 56, 330–338. [Google Scholar] [CrossRef]
- Ballan, M.; Bovo, S.; Schiavo, G.; Schiavitto, M.; Negrini, R.; Fontanesi, L. Genomic diversity and signatures of selection in meat and fancy rabbit breeds based on high-density marker data. Genet. Sel. Evol. 2022, 54, 3. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Ribani, A.; Moscatelli, G.; Bonacini, M.; Prandi, M.; Mancin, E.; Mantovani, R.; Dall’Olio, S.; Fontanesi, L. Comparative analysis of inbreeding parameters and runs of homozygosity islands in 2 Italian autochthonous cattle breeds mainly raised in the Parmigiano-Reggiano cheese production region. J. Dairy Sci. 2022, 105, 2408–2425. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wang, K.; Zhou, J.; Yang, Q.; Yang, X.; Jiang, A.; Jiang, Y.; Li, M.; Zhu, L.; Bai, L.; et al. A genome wide association study for the number of animals born dead in domestic pigs. BMC Genet. 2019, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Deng, F.; Wu, Z.; Chen, S.-Y.; Shi, Y.; Jia, X.; Hu, S.; Wang, J.; Cao, W.; Lai, S.-J. A Genome-Wide Association Study Identifying Genetic Variants Associated with Growth, Carcass and Meat Quality Traits in Rabbits. Animals 2020, 10, 1068. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, Z.; Glória, L.S.; Zhang, K.; Zhang, C.; Yang, R.; Luo, X.; Jia, X.; Lai, S.J.; Chen, S.Y. Genome-Wide Association Studies for Growth Curves in Meat Rabbits Through the Single-Step Nonlinear Mixed Model. Front. Genet. 2021, 12, 750939. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Bovo, S.; Bertolini, F.; Tinarelli, S.; Dall’Olio, S.; Nanni Costa, L.; Gallo, M.; Fontanesi, L. Comparative evaluation of genomic inbreeding parameters in seven commercial and autochthonous pig breeds. Animal 2020, 14, 910–920. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Rohrer, G.A.; Nonneman, D.J.; Wiedmann, R.T.; Schneider, J.F. A study of vertebra number in pigs confirms the association of vertnin and reveals additional QTL. BMC Genet. 2015, 16, 129. [Google Scholar] [CrossRef]
- Akanno, E.C.; Chen, L.; Abo-Ismail, M.K.; Crowley, J.J.; Wang, Z.; Li, C.; Basarab, J.A.; MacNeil, M.D.; Plastow, G.S. Genome-wide association scan for heterotic quantitative trait loci in multi-breed and crossbred beef cattle. Genet. Sel. Evol. 2018, 50, 48. [Google Scholar] [CrossRef] [PubMed]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015, 43, W589–W598. [Google Scholar] [CrossRef] [PubMed]
- Teo, Y.Y. Common statistical issues in genome-wide association studies: A review on power, data quality control, genotype calling and population structure. Curr. Opin. Lipidol. 2008, 19, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhang, S.; Zhang, H.; Zhang, Z.; Chen, N.; Li, Z.; Sun, H.; Liu, X.; Lyu, S.; Wang, X.; et al. Assessing genomic diversity and signatures of selection in Jiaxian Red cattle using whole-genome sequencing data. BMC Genom. 2021, 22, 43. [Google Scholar] [CrossRef] [PubMed]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Qu, K.; Ma, X.; Hanif, Q.; Zhang, J.; Liu, J.; Chen, N.; Suolang, Q.; Lei, C.; Huang, B. Whole-Genome Analyses Reveal Genomic Characteristics and Selection Signatures of Lincang Humped Cattle at the China-Myanmar Border. Front. Genet. 2022, 13, 833503. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Qu, K.; Hanif, Q.; Zhang, J.; Liu, J.; Chen, N.; Suolang, Q.; Lei, C.; Huang, B. Whole-Genome Sequencing of Endangered Dengchuan Cattle Reveals Its Genomic Diversity and Selection Signatures. Front. Genet. 2022, 13, 833475. [Google Scholar] [CrossRef]
- Charlesworth, D.; Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 2009, 10, 783–796. [Google Scholar] [CrossRef]
- Bjelland, D.W.; Weigel, K.A.; Vukasinovic, N.; Nkrumah, J.D. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J. Dairy Sci. 2013, 96, 4697–4706. [Google Scholar] [CrossRef]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Yao, Z.; Zhang, S.; Wang, X.; Guo, Y.; Xin, X.; Zhang, Z.; Xu, Z.; Wang, E.; Jiang, Y.; Huang, Y. Genetic diversity and signatures of selection in BoHuai goat revealed by whole-genome sequencing. BMC Genom. 2023, 24, 116. [Google Scholar] [CrossRef] [PubMed]
- Addo, S.; Klingel, S.; Thaller, G.; Hinrichs, D. Genetic diversity and the application of runs of homozygosity-based methods for inbreeding estimation in German White-headed Mutton sheep. PLoS ONE 2021, 16, e0250608. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Kostetskii, I.; Radice, G.L. N-cadherin is not essential for limb mesenchymal chondrogenesis. Dev. Dyn. 2005, 232, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Jahuey-Martínez, F.J.; Parra-Bracamonte, G.M.; Sifuentes-Rincón, A.M.; Martínez-González, J.C.; Gondro, C.; García-Pérez, C.A.; López-Bustamante, L.A. Genomewide association analysis of growth traits in Charolais beef cattle. J. Anim. Sci. 2016, 94, 4570–4582. [Google Scholar] [CrossRef]
- Yang, C.; Guo, X.; Dong, F.; Meng, F.; Wang, L.; Wang, P.; Zhang, C.; Ren, Y. miR-542-3p reduces antioxidant capacity in goat caput epididymal epithelial cells by targeting glutathione peroxidase 5 (GPx5). Theriogenology 2022, 186, 168–174. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DA | N | Max1 (g) | Min1 (g) | M1 (g) | SD (g) |
|---|---|---|---|---|---|
| 35 | 378 | 1095 | 456 | 786.31 | 120.42 |
| 42 | 379 | 1414 | 665 | 1011.42 | 114.37 |
| 49 | 380 | 1657 | 745 | 1243.56 | 138.14 |
| 56 | 380 | 2005 | 826 | 1473.96 | 182.26 |
| 63 | 360 | 2354 | 906 | 1706.34 | 238.28 |
| 70 | 352 | 2726 | 930 | 1943.69 | 295.90 |
| 84 | 339 | 2750 | 1080 | 2136.16 | 280.64 |
| DA | N | Max2 (%) | Min2 (%) | M2 (%) | SD (%) |
|---|---|---|---|---|---|
| 35 | 378 | 0.485 | −0.378 | −0.006 | 0.176 |
| 42 | 379 | 0.418 | −0.323 | −0.012 | 0.133 |
| 49 | 380 | 0.259 | −0.492 | −0.119 | 0.124 |
| 56 | 378 | 0.463 | −0.506 | −0.055 | 0.146 |
| 63 | 358 | 0.514 | −0.517 | −0.016 | 0.155 |
| 70 | 345 | 0.370 | −0.548 | −0.011 | 0.161 |
| 84 | 332 | 0.306 | −0.523 | −0.017 | 0.139 |
| Character | 35~42 DA | 42~49 DA | 49~56 DA | 56~63 DA | 63~70 DA | 70~84 DA |
|---|---|---|---|---|---|---|
| DG | 31.91 ± 10.63 | 32.99 ± 9.21 | 32.91 ± 9.35 | 32.91 ± 9.35 | 33.02 ± 9.56 | 12.06 ± 23.95 |
| HW | 0.001 ± 0.39 | −0.37 ± 0.28 | −0.39 ± 4.23 | 0.78 ± 2.16 | 0.14 ± 0.59 | −0.13 ± 2.32 |
| Chromosomes | N | AD (bp) | MAF |
|---|---|---|---|
| 1 | 6966 | 27.966 | 0.264 |
| 2 | 6654 | 26.137 | 0.257 |
| 3 | 5894 | 26.383 | 0.251 |
| 4 | 3205 | 28.495 | 0.238 |
| 5 | 1195 | 31.161 | 0.268 |
| 6 | 817 | 33.428 | 0.239 |
| 7 | 6752 | 25.705 | 0.249 |
| 8 | 4714 | 23.620 | 0.252 |
| 9 | 4613 | 25.090 | 0.254 |
| 10 | 1769 | 25.160 | 0.248 |
| 11 | 3278 | 26.673 | 0.248 |
| 12 | 5669 | 27.375 | 0.252 |
| 13 | 4849 | 29.524 | 0.261 |
| 14 | 6000 | 27.300 | 0.259 |
| 15 | 4517 | 23.464 | 0.252 |
| 16 | 2809 | 30.064 | 0.259 |
| 17 | 3207 | 26.484 | 0.269 |
| 18 | 2450 | 27.053 | 0.249 |
| 19 | 1748 | 30.828 | 0.249 |
| 20 | 1178 | 25.569 | 0.243 |
| 21 | 295 | 42.013 | 0.244 |
| Chromosomes | Samples | ROHs | Qty | Avg (Mb) | FROH |
|---|---|---|---|---|---|
| 1 | 380 | 3878 | 171 | 1.24 ± 0.46 | 7.01 × 10−8 |
| 2 | 380 | 3567 | 193 | 1.32 ± 0.51 | 8.04 × 10−8 |
| 3 | 379 | 3257 | 91 | 1.22 ± 0.43 | 8.83 × 10−8 |
| 4 | 378 | 2223 | 149 | 1.36 ± 0.63 | 1.92 × 10−7 |
| 5 | 209 | 443 | 0 | 1.06 ± 0.27 | 1.90 × 10−7 |
| 6 | 255 | 385 | 17 | 1.50 ± 0.44 | 5.22 × 10−7 |
| 7 | 380 | 4065 | 281 | 1.27 ± 0.66 | 8.73 × 10−8 |
| 8 | 379 | 2489 | 38 | 1.22 ± 0.41 | 1.13 × 10−7 |
| 9 | 381 | 2781 | 197 | 1.35 ± 0.56 | 1.29 × 10−7 |
| 10 | 326 | 921 | 59 | 1.20 ± 0.55 | 2.41 × 10−7 |
| 11 | 377 | 1638 | 2 | 1.15 ± 0.37 | 1.33 × 10−7 |
| 12 | 380 | 2539 | 101 | 1.23 ± 0.46 | 7.03 × 10−8 |
| 13 | 375 | 2090 | 132 | 1.38 ± 0.51 | 8.73 × 10−8 |
| 14 | 380 | 3221 | 79 | 1.19 ± 0.41 | 7.32 × 10−8 |
| 15 | 380 | 3009 | 119 | 1.25 ± 0.51 | 1.47 × 10−7 |
| 16 | 353 | 1109 | 124 | 1.31 ± 0.63 | 1.16 × 10−7 |
| 17 | 379 | 1835 | 111 | 1.32 ± 0.57 | 1.73 × 10−7 |
| 18 | 358 | 1131 | 0 | 1.00 ± 0.28 | 1.24 × 10−7 |
| 19 | 349 | 849 | 56 | 1.28 ± 0.54 | 2.05 × 10−7 |
| 20 | 269 | 501 | 3 | 1.25 ± 0.38 | 2.82 × 10−7 |
| 21 | 87 | 87 | 0 | 0.89 ± 0.05 | 2.75 × 10−7 |
| Chromosome | Significant SNPs | Position (bp) | Candidate Genes |
|---|---|---|---|
| 1 | ENSOCUG00000014693 | 115177017 | PGR |
| 2 | ENSOCUG00000002594 | 62292147 | SLC25A4 |
| ENSOCUG00000011227 | 135606186 | LHCGR | |
| ENSOCUG00000010418 | 49018842 | SAP30 | |
| ENSOCUG00000009784 | 135741577 | FOXN2 | |
| 4 | ENSOCUG00000017314 | 63287210 | MGAT4C |
| ENSOCUG00000005909 | 44797807 | HELB | |
| 6 | ENSOCUG00000001191 | 11540770 | LYRM1 |
| 7 | ENSOCUG00000011449 | 19355249 | SPAM1 |
| ENSOCUG00000012455 | 29470957 | MDFIC | |
| ENSOCUG00000000303 | 29470957 | PPP1R3A | |
| 9 | ENSOCUG00000013743 | 72501512 | MEP1B |
| 10 | ENSOCUG00000015979 | 16510779 | DPY19L1 |
| 13 | ENSOCUG00000012320 | 44937297 | TBX15 |
| 14 | ENSOCUG00000001739 | 129651182 | CADM2 |
| 15 | ENSOCUG00000008959 | 94164303 | MYOZ2 |
| ENSOCUG00000001300 | 57092972 | CCSER1 | |
| 16 | ENSOCUG00000006634 | 17521122 | FAM107B |
| 17 | ENSOCUG00000011430 | 49383678 | AP4S1 |
| 19 | ENSOCUG00000007606 | 52334275 | MAP2K6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Z.; Li, Y.; Yang, L.; Cui, M.; Wang, Z.; Sun, W.; Wang, J.; Chen, S.; Lai, S.; Jia, X. Genome-Wide Association Studies of Growth Trait Heterosis in Crossbred Meat Rabbits. Animals 2024, 14, 2096. https://doi.org/10.3390/ani14142096
Xiao Z, Li Y, Yang L, Cui M, Wang Z, Sun W, Wang J, Chen S, Lai S, Jia X. Genome-Wide Association Studies of Growth Trait Heterosis in Crossbred Meat Rabbits. Animals. 2024; 14(14):2096. https://doi.org/10.3390/ani14142096
Chicago/Turabian StyleXiao, Zhanjun, Yuchao Li, Li Yang, Mingyan Cui, Zicheng Wang, Wenqiang Sun, Jie Wang, Shiyi Chen, Songjia Lai, and Xianbo Jia. 2024. "Genome-Wide Association Studies of Growth Trait Heterosis in Crossbred Meat Rabbits" Animals 14, no. 14: 2096. https://doi.org/10.3390/ani14142096
APA StyleXiao, Z., Li, Y., Yang, L., Cui, M., Wang, Z., Sun, W., Wang, J., Chen, S., Lai, S., & Jia, X. (2024). Genome-Wide Association Studies of Growth Trait Heterosis in Crossbred Meat Rabbits. Animals, 14(14), 2096. https://doi.org/10.3390/ani14142096