
A 16S RNA Analysis of Yangzhou Geese with Varying Body Weights: Gut Microbial Difference and Its Correlation with Body Weight Parameters

 and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. DNA Extraction and 16S rRNA Sequencing
2.3. Analysis of 16S rRNA Sequencing Data
2.4. Statistical Analysis
3. Results
3.1. Analysis of Growth Performance in L and S Groups of Yangzhou Geese
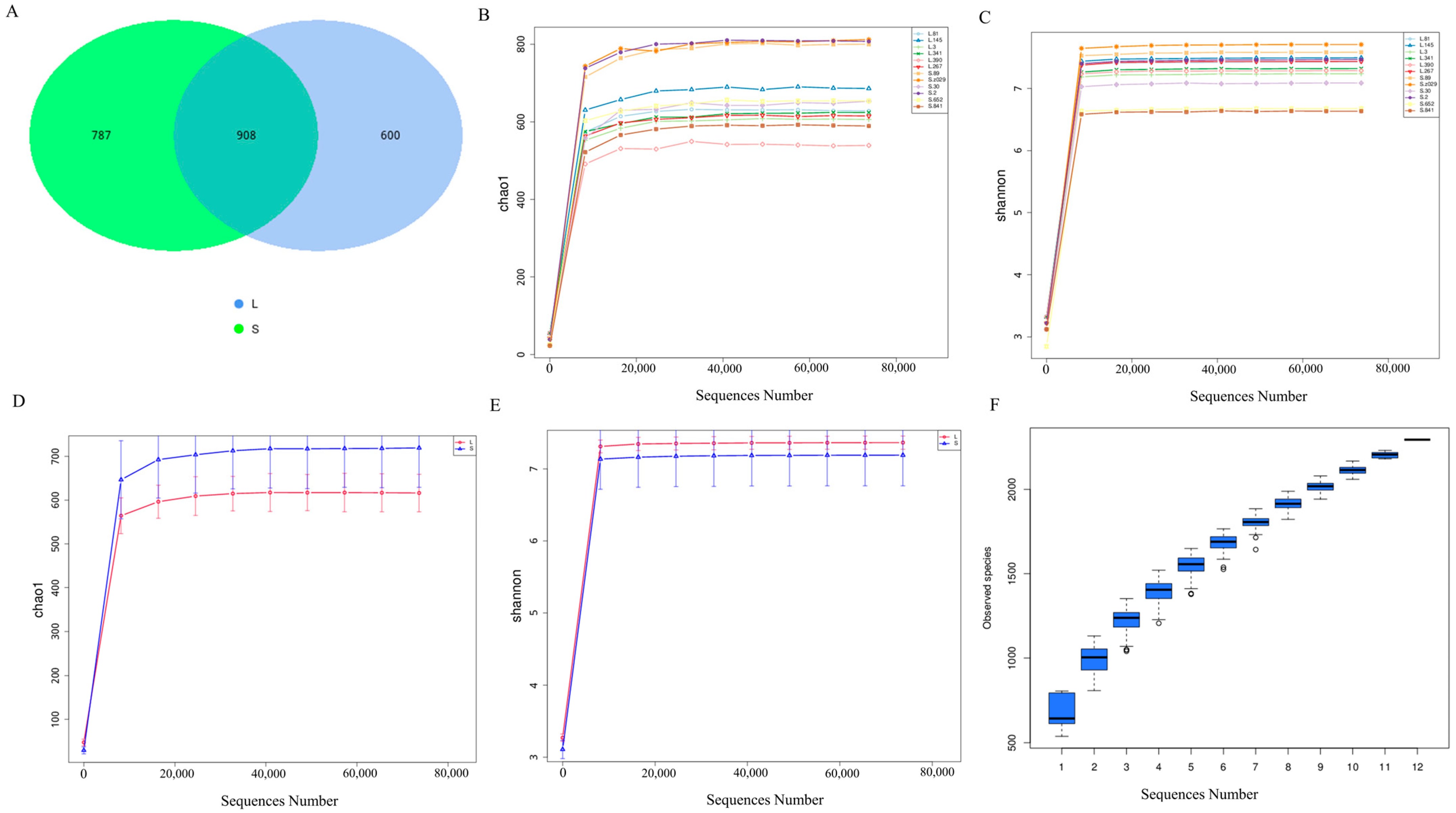
3.2. Overview of Cecal Microbial Data in Groups L and S
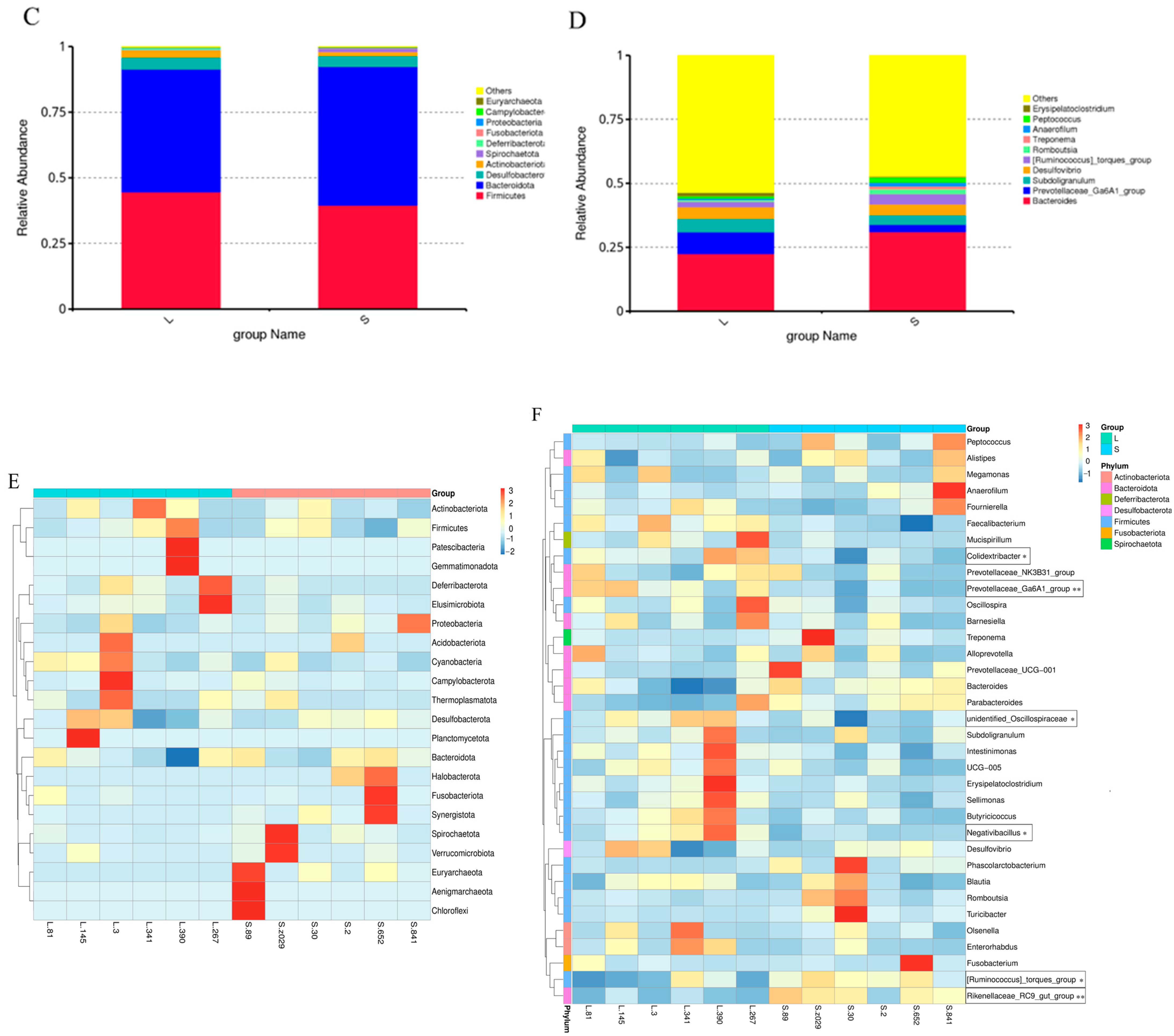
3.3. Analysis of Cecal Microbial Diversity and Composition in Groups L and S
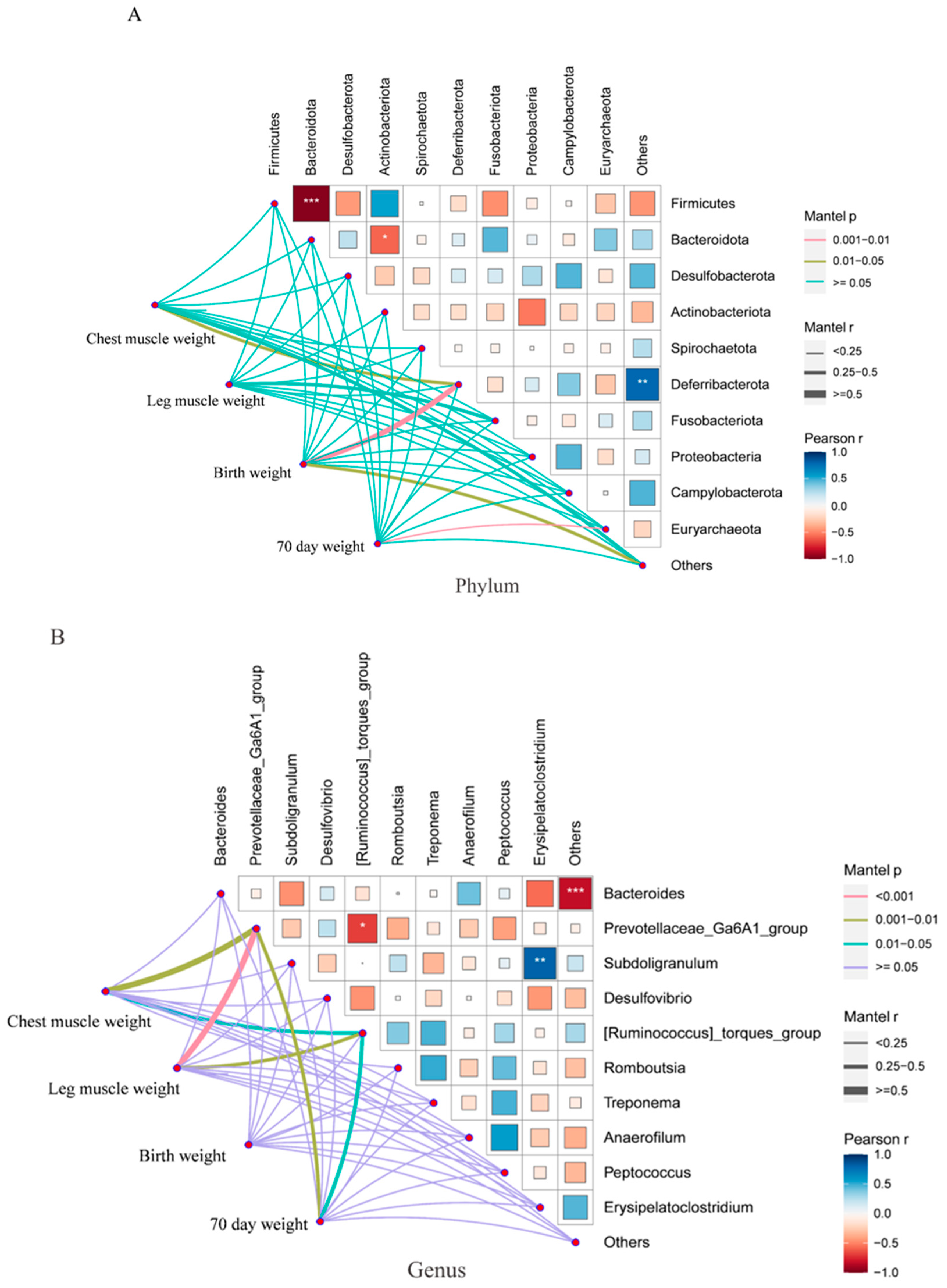
3.4. Correlation Analysis of Cecal Microbiota and Weight-Related Traits in Groups L and S
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abd El-Hack, M.E.; El-Saadony, M.T.; Alqhtani, A.H.; Swelum, A.A.; Salem, H.M.; Elbestawy, A.R.; Noreldin, A.E.; Babalghith, A.O.; Khafaga, A.F.; Hassan, M.I.; et al. The relationship among avian influenza, gut microbiota and chicken immunity: An updated overview. Poult. Sci. 2022, 101, 102021. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, C.; Simujide, H.; Aricha, H.; Zhang, J.; Liu, B.; Aorigele, C. Effects of Pathogenic Escherichia coli Infection on the Flora Composition, Function, and Content of Short-Chain Fatty Acids in Calf Feces. Animals 2022, 12, 959. [Google Scholar] [CrossRef] [PubMed]
- Unterweger, C.; Kahler, A.; Gerlach, G.F.; Viehmann, M.; von Altrock, A.; Hennig-Pauka, I. Administration of non-pathogenic isolates of Escherichia coli and Clostridium perfringens type A to piglets in a herd affected with a high incidence of neonatal diarrhoea. Anim. Int. J. Anim. Biosci. 2017, 11, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Wu, Q.F.; Ma, S.C.; Wang, J.M.; Wei, B.; Xi, Y.; Han, C.C.; Li, L.; He, H.; Liu, H.H. Effect of feed restriction on the intestinal microbial community structure of growing ducks. Arch. Microbiol. 2021, 204, 85. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; O’Riordan, M.X. Regulation of bacterial pathogenesis by intestinal short-chain Fatty acids. Adv. Appl. Microbiol. 2013, 85, 93–118. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.H.; Lin, S.Y.; Chen, L.L.; Ouyang, K.H.; Wang, W.J. The Interaction between Flavonoids and Intestinal Microbes: A Review. Foods 2023, 12, 320. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wu, H.; Lv, Y.; You, H.; Zha, L.; Li, Q.; Huang, Y.; Tian, J.; Chen, Q.; Shen, Y.; et al. Gastrointestinal Development and Microbiota Responses of Geese to Honeycomb Flavonoids Supplementation. Front. Vet. Sci. 2021, 8, 739237. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Hemarajata, P.; Versalovic, J. Effects of probiotics on gut microbiota: Mechanisms of intestinal immunomodulation and neuromodulation. Ther. Adv. Gastroenterol. 2013, 6, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Leng, X.; Zhao, Y.; Zhao, Y.; Wang, Q. Effects of dietary Artemisia annua supplementation on growth performance, antioxidant capacity, immune function, and gut microbiota of geese. Poult. Sci. 2024, 103, 103594. [Google Scholar] [CrossRef]
- Hou, Q.; Qian, Z.; Wu, P.; Shen, M.; Li, L.; Zhao, W. 1-Deoxynojirimycin from mulberry leaves changes gut digestion and microbiota composition in geese. Poult. Sci. 2020, 99, 5858–5866. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, X.; Liu, Y.; Gong, S.; Yang, Y.; Wang, C.; Wang, H.; He, D. Bile acids supplementation modulates lipid metabolism, intestinal function, and cecal microbiota in geese. Front. Microbiol. 2023, 14, 1185218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, N.; Wu, J.; Song, L.; Bao, Q.; Weng, K.; Zhang, Y.; Vongsangnak, W.; Chen, G.; Xu, Q. Gut microbiome and serum metabolome analyses identify Bacteroides fragilis as regulators of serotonin content and PRL secretion in broody geese. J. Integr. Agric. 2024, 23, 2033–2051. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Zhang, Y.; Ni, H.; Xiao, Q.; Yin, Y.; Ren, J.; Zhao, P.; Zhang, Z.; Li, X.; Li, Y.; et al. Optimization of Fermented Maize Stover for the Fattening Phase of Geese: Effect on Production Performance and Gut Microflora. Animals 2024, 14, 433. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Li, D.; Zhou, B.; Jiang, Y.; Bai, H.; Zhang, Y.; Xu, Q.; Zhao, W.; Chen, G. Comparative characterization of bacterial communities in geese consuming of different proportions of ryegrass. PLoS ONE 2019, 14, e0223445. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sheng, H.F.; He, Y.; Wu, J.Y.; Jiang, Y.X.; Tam, N.F.; Zhou, H.W. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl. Environ. Microbiol. 2012, 78, 8264–8271. [Google Scholar] [CrossRef] [PubMed]
- Konstantinov, S.R.; Favier, C.F.; Zhu, W.; Williams, B.A.; Klüß, J.; Souffrant, W.B.; Vos, W.M.d.; Akkermans, A.D.L.; Smidt, H.J.A.R. Microbial diversity studies of the porcine gastrointestinal ecosystem during weaning transition. Anim. Res. 2004, 53, 317–324. [Google Scholar] [CrossRef]
- Li, Y.; Yang, H.; Xu, L.; Wang, Z.; Zhao, Y.; Chen, X. Effects of dietary fiber levels on cecal microbiota composition in geese. Asian-Australas J. Anim. Sci. 2018, 31, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- NY/T 823-2020; Terminology and Measurement Methods for Poultry Production Performance. Ministry of Agriculture and Rural Affairs of the People’s Republic of China: Beijing, China, 2020.
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Equbal, J.; Lakra, R.K.; Savurirajan, M.; Satyam, K.; Thiruchitrambalam, G. Assessing the benthic quality status of three fine sand tropical beaches from the Andaman Islands through the application of marine biotic indices. Environ. Monit. Assess. 2022, 194, 479. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Chen, L.; Bai, X.; Wang, T.; Liu, J.; Miao, X.; Zeng, B.; Li, D. Gut Microbial Diversity Analysis of Different Native Chickens and Screening of Chicken-Derived Probiotics. Animals 2023, 13, 3672. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Zhang, M.; Liu, J.; Zhu, W. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: Membership and potential function. Sci. Rep. 2015, 5, 16116. [Google Scholar] [CrossRef]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarăes, V.; Sokol, H.; Doré, J.; Corthier, G.; Furet, J.P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef]
- Zafar, H.; Saier, M.H., Jr. Gut Bacteroides species in health and disease. Gut Microbes 2021, 13, 1848158. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Scheppach, W.; Luehrs, H.; Menzel, T. Beneficial health effects of low-digestible carbohydrate consumption. Br. J. Nutr. 2001, 85 (Suppl. S1), S23–S30. [Google Scholar] [CrossRef]
- von Rosenvinge, E.C.; Song, Y.; White, J.R.; Maddox, C.; Blanchard, T.; Fricke, W.F. Immune status, antibiotic medication and pH are associated with changes in the stomach fluid microbiota. ISME J. 2013, 7, 1354–1366. [Google Scholar] [CrossRef]
- Ma, L.; Lyu, W.; Zeng, T.; Wang, W.; Chen, Q.; Zhao, J.; Zhang, G.; Lu, L.; Yang, H.; Xiao, Y. Duck gut metagenome reveals the microbiome signatures linked to intestinal regional, temporal development, and rearing condition. iMeta 2024, e198. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | S Group | L Group |
|---|---|---|
| Heart weight (g) | 26.32 ± 2.82 | 23.64 ± 4.07 |
| Liver weight (g) | 70.26 ± 12.75 | 65.24 ± 9.45 |
| Spleen weight (g) | 5.57 ± 1.79 | 3.93 ± 0.64 |
| Gizzard net weight (g) | 159.17 ± 17.41 | 148.67 ± 15.18 |
| Abdominal fat weight (g) | 60.11 ± 23.16 | 41.21 ± 9.30 |
| Leg muscle weight (g) | 342.7 ± 15.77 b | 377.6 ± 19.61 a |
| Breast muscle weight (g) | 233.7 ± 21.50 b | 278 ± 32.26 a |
| Birth weight (g) | 95.67 ± 4.11 b | 112.17 ± 11.33 a |
| 70-day-old weight (g) | 2841.67 ± 72.90 B | 4390 ± 130.51 A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Fan, S.; Wu, H.; Li, H.; Shan, X.; Wang, M.; Zhang, Y.; Xu, Q.; Chen, G. A 16S RNA Analysis of Yangzhou Geese with Varying Body Weights: Gut Microbial Difference and Its Correlation with Body Weight Parameters. Animals 2024, 14, 2042. https://doi.org/10.3390/ani14142042
Xu X, Fan S, Wu H, Li H, Shan X, Wang M, Zhang Y, Xu Q, Chen G. A 16S RNA Analysis of Yangzhou Geese with Varying Body Weights: Gut Microbial Difference and Its Correlation with Body Weight Parameters. Animals. 2024; 14(14):2042. https://doi.org/10.3390/ani14142042
Chicago/Turabian StyleXu, Xinlei, Suyu Fan, Hao Wu, Haoyu Li, Xiaoyu Shan, Mingfeng Wang, Yang Zhang, Qi Xu, and Guohong Chen. 2024. "A 16S RNA Analysis of Yangzhou Geese with Varying Body Weights: Gut Microbial Difference and Its Correlation with Body Weight Parameters" Animals 14, no. 14: 2042. https://doi.org/10.3390/ani14142042
APA StyleXu, X., Fan, S., Wu, H., Li, H., Shan, X., Wang, M., Zhang, Y., Xu, Q., & Chen, G. (2024). A 16S RNA Analysis of Yangzhou Geese with Varying Body Weights: Gut Microbial Difference and Its Correlation with Body Weight Parameters. Animals, 14(14), 2042. https://doi.org/10.3390/ani14142042