

Application of Host-Depleted Nanopore Metagenomic Sequencing in the Clinical Detection of Pathogens in Pigs and Cats
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
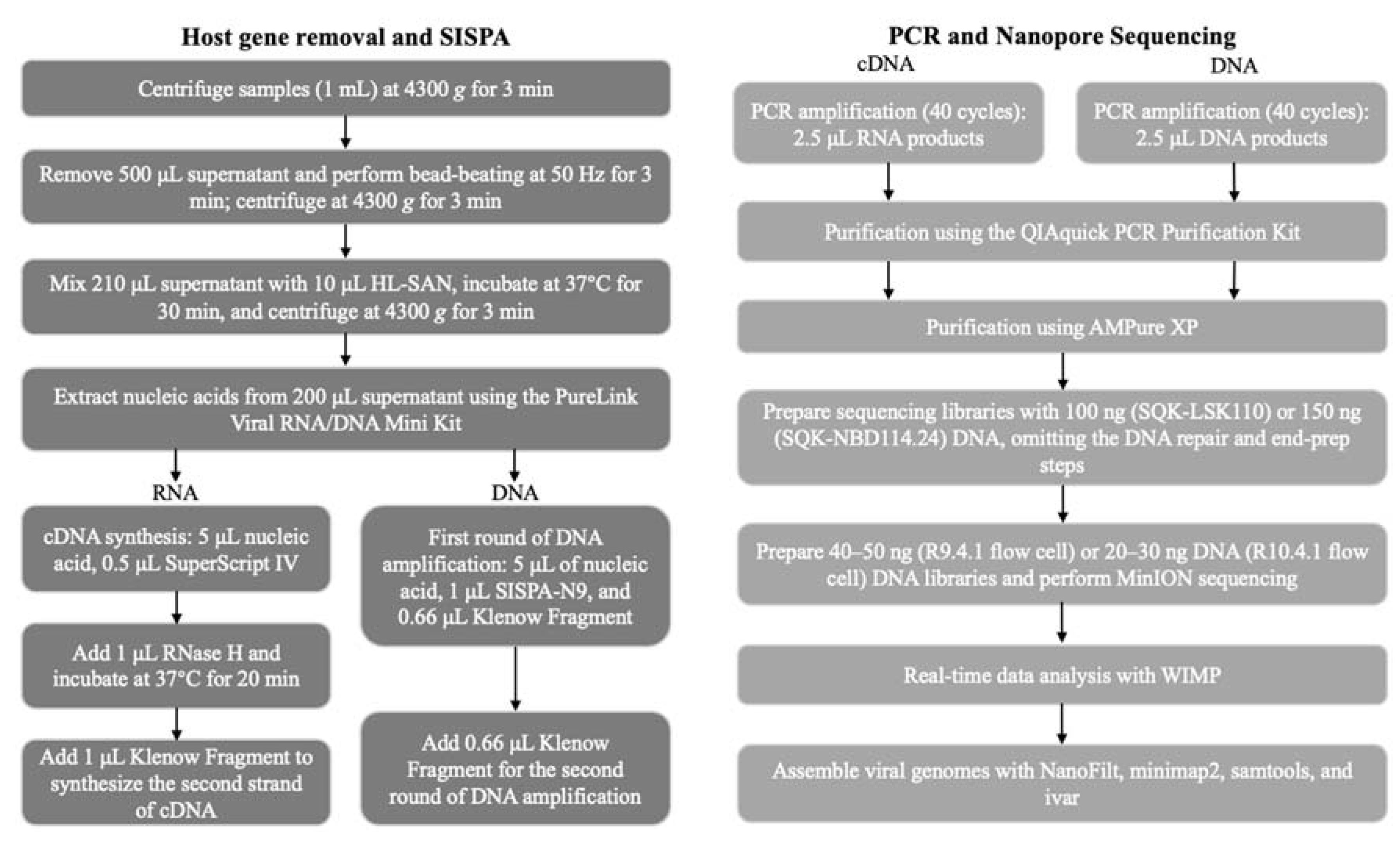
2.1. Sample Information
2.2. Metagenomic Sequencing
2.2.1. Nuclease Digestion and Nucleic Acid Extraction
2.2.2. RNA Reverse Transcription and Second-Strand Synthesis
2.2.3. Addition of Random Primers to Both Ends of DNA
2.2.4. PCR Amplification
2.2.5. PCR Product Purification
2.2.6. Preparation of Nanopore Sequencing Libraries
2.2.7. Sequencing and Data Analysis
3. Results
3.1. Clinical Sample Detection Results
3.2. Host Gene Removal Efficiency
3.3. Sequencing Data Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, N.; Cao, J.; Xu, J.; Liu, B.; Liu, B.; Chen, D.; Xia, B.; Chen, L.; Zhang, W.; Zhang, Y.; et al. Targeting RNA with Next- and Third-Generation Sequencing Improves Pathogen Identification in Clinical Samples. Adv. Sci. 2021, 8, e2102593. [Google Scholar] [CrossRef] [PubMed]
- Donati, C.; Zolfo, M.; Albanese, D.; Tin Truong, D.; Asnicar, F.; Iebba, V.; Cavalieri, D.; Jousson, O.; De Filippo, C.; Huttenhower, C.; et al. Uncovering oral Neisseria tropism and persistence using metagenomic sequencing. Nat. Microbiol. 2016, 1, 16070. [Google Scholar] [CrossRef] [PubMed]
- Zinter, M.S.; Dvorak, C.C.; Mayday, M.Y.; Iwanaga, K.; Ly, N.P.; McGarry, M.E.; Church, G.D.; Faricy, L.E.; Rowan, C.M.; Hume, J.R.; et al. Pulmonary Metagenomic Sequencing Suggests Missed Infections in Immunocompromised Children. Clin. Infect. Dis. 2019, 68, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Phelan, A.L.; Katz, R.; Gostin, L.O. The Novel Coronavirus Originating in Wuhan, China: Challenges for Global Health Governance. JAMA 2020, 323, 709–710. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, W.; Zhao, X.; Zai, J.; Zhao, Q.; Li, Y.; Chaillon, A. Transmission dynamics and evolutionary history of 2019-nCoV. J. Med. Virol. 2020, 92, 501–511. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Xie, C.; Ding, S.; Yang, H.; Guo, S.; Li, J.; Qin, L.; Ban, F.; Wang, D.; et al. A Novel Human Acute Encephalitis Caused by Pseudorabies Virus Variant Strain. Clin. Infect. Dis. 2021, 73, e3690–e3700. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Schuele, L.; Lizarazo-Forero, E.; Strutzberg-Minder, K.; Schutze, S.; Lobert, S.; Lambrecht, C.; Harlizius, J.; Friedrich, A.W.; Peter, S.; Rossen, J.W.A.; et al. Application of shotgun metagenomics sequencing and targeted sequence capture to detect circulating porcine viruses in the Dutch-German border region. Transbound Emerg. Dis. 2022, 69, 2306–2319. [Google Scholar] [CrossRef]
- Hoon-Hanks, L.L.; McGrath, S.; Tyler, K.L.; Owen, C.; Stenglein, M.D. Metagenomic Investigation of Idiopathic Meningoencephalomyelitis in Dogs. J. Vet. Intern. Med. 2018, 32, 324–330. [Google Scholar] [CrossRef]
- Akacin, I.; Ersoy, S.; Doluca, O.; Gungormusler, M. Comparing the significance of the utilization of next generation and third generation sequencing technologies in microbial metagenomics. Microbiol. Res. 2022, 264, 127154. [Google Scholar] [CrossRef]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-Generation Sequencing in the Clinical Laboratory: Exploring the Advantages and Challenges of Nanopore Sequencing. J. Clin. Microbiol. 2019, 58, e01315-19. [Google Scholar] [CrossRef] [PubMed]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 2019, 37, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, G.; Lau, H.C.; Yu, J. Metagenomic Sequencing for Microbial DNA in Human Samples: Emerging Technological Advances. Int. J. Mol. Sci. 2022, 23, 2181. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, K.M.; Erb-Downward, J.R.; Bao, Y.; Branton, W.R.; Falkowski, N.R.; Newton, D.W.; Huffnagle, G.B.; Dickson, R.P. Rapid Pathogen Identification in Bacterial Pneumonia Using Real-Time Metagenomics. Am. J. Respir. Crit. Care Med. 2017, 196, 1610–1612. [Google Scholar] [CrossRef]
- Gil, P.; Dupuy, V.; Koual, R.; Exbrayat, A.; Loire, E.; Fall, A.G.; Gimonneau, G.; Biteye, B.; Talla Seck, M.; Rakotoarivony, I.; et al. A library preparation optimized for metagenomics of RNA viruses. Mol. Ecol. Resour. 2021, 21, 1788–1807. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Monteil-Bouchard, S.; Robert, C.; Pascalis, H.; Michelle, C.; Jardot, P.; Charrel, R.; Raoult, D.; Desnues, C. Host-Associated Metagenomics: A Guide to Generating Infectious RNA Viromes. PLoS ONE 2015, 10, e0139810. [Google Scholar] [CrossRef]
- Street, T.L.; Barker, L.; Sanderson, N.D.; Kavanagh, J.; Hoosdally, S.; Cole, K.; Newnham, R.; Selvaratnam, M.; Andersson, M.; Llewelyn, M.J.; et al. Optimizing DNA Extraction Methods for Nanopore Sequencing of Neisseria gonorrhoeae Directly from Urine Samples. J. Clin. Microbiol. 2020, 58, e01822-19. [Google Scholar] [CrossRef]
- Simner, P.J.; Miller, H.B.; Breitwieser, F.P.; Pinilla Monsalve, G.; Pardo, C.A.; Salzberg, S.L.; Sears, C.L.; Thomas, D.L.; Eberhart, C.G.; Carroll, K.C. Development and Optimization of Metagenomic Next-Generation Sequencing Methods for Cerebrospinal Fluid Diagnostics. J. Clin. Microbiol. 2018, 56, e00472-18. [Google Scholar] [CrossRef]
- Charalampous, T.; Alcolea-Medina, A.; Snell, L.B.; Williams, T.G.S.; Batra, R.; Alder, C.; Telatin, A.; Camporota, L.; Meadows, C.I.S.; Wyncoll, D.; et al. Evaluating the potential for respiratory metagenomics to improve treatment of secondary infection and detection of nosocomial transmission on expanded COVID-19 intensive care units. Genome Med. 2021, 13, 182. [Google Scholar] [CrossRef]
- Hellmann, K.T.; Tuura, C.E.; Fish, J.; Patel, J.M.; Robinson, D.A. Viability-Resolved Metagenomics Reveals Antagonistic Colonization Dynamics of Staphylococcus epidermidis Strains on Preterm Infant Skin. mSphere 2021, 6, e0053821. [Google Scholar] [CrossRef]
- Marotz, C.A.; Sanders, J.G.; Zuniga, C.; Zaramela, L.S.; Knight, R.; Zengler, K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 2018, 6, 42. [Google Scholar] [CrossRef]
- Rubiola, S.; Chiesa, F.; Dalmasso, A.; Di Ciccio, P.; Civera, T. Detection of Antimicrobial Resistance Genes in the Milk Production Environment: Impact of Host DNA and Sequencing Depth. Front. Microbiol. 2020, 11, 1983. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. Rapid Sequencing DNA—Viral Metagenomics for Respiratory Samples and Skin Lesion Swabs (SQK-RPB004) Protocol. Available online: https://community.nanoporetech.com/docs/prepare/library_prep_protocols/viral-metagenomics-RNA-DNA/v/vmm_9160_v1_revf_16jun2022 (accessed on 1 November 2023).
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J.; et al. Metagenomic Nanopore Sequencing of Influenza Virus Direct from Clinical Respiratory Samples. J. Clin. Microbiol. 2019, 58, e00963-19. [Google Scholar] [CrossRef]
- Israeli, O.; Guedj-Dana, Y.; Shifman, O.; Lazar, S.; Cohen-Gihon, I.; Amit, S.; Ben-Ami, R.; Paran, N.; Schuster, O.; Weiss, S.; et al. Rapid Amplicon Nanopore Sequencing (RANS) for the Differential Diagnosis of Monkeypox Virus and Other Vesicle-Forming Pathogens. Viruses 2022, 14, 1817. [Google Scholar] [CrossRef] [PubMed]
- Schuele, L.; Cassidy, H.; Lizarazo, E.; Strutzberg-Minder, K.; Schuetze, S.; Loebert, S.; Lambrecht, C.; Harlizius, J.; Friedrich, A.W.; Peter, S.; et al. Assessment of Viral Targeted Sequence Capture Using Nanopore Sequencing Directly from Clinical Samples. Viruses 2020, 12, 1358. [Google Scholar] [CrossRef] [PubMed]
- Radzieta, M.; Sadeghpour-Heravi, F.; Peters, T.J.; Hu, H.; Vickery, K.; Jeffries, T.; Dickson, H.G.; Schwarzer, S.; Jensen, S.O.; Malone, M. A multiomics approach to identify host-microbe alterations associated with infection severity in diabetic foot infections: A pilot study. npj Biofilms Microbiomes 2021, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Sample No. | Sample Type | Clinical Symptoms | Detection Items |
|---|---|---|---|
| 1 | Cat Serum | Depression, hind limb weakness, decreased neutrophils, monocytes, eosinophils, and platelets | PCR 1: FPV, FHV-1, FCV, FCoV, FIV, FeLV Metagenomic Sequencing |
| 2 | Cat Nasopharyngeal Swab + Feces | Fever, increased nasal and ocular secretions, diarrhea | PCR 1: Chlamydophila felis, Mycoplasma felis, FPV, FCV, FHV-1 Metagenomic Sequencing |
| 3 | Pig Lung Tissue | Respiratory symptoms in sows, sudden death | PCR 1: CSFV, PRRSV, PCV2, PRV, ASFV Bacterial Culture 2 + 16S rRNA Sequencing 3 Metagenomic Sequencing |
| 4 | Cat Nasopharyngeal Swab | Respiratory symptoms | PCR 1: FCV, FHV-1, Chlamydophila felis, Mycoplasma felis, Bordetella bronchiseptica Metagenomic Sequencing |
| 5 | Pig Lung Tissue Pool (4 pigs) | Severe respiratory symptoms, fever, and high mortality in nursery pigs | PCR 1: CSFV, PRRSV, PRV, PCV2 Bacterial Culture 2 + 16S rRNA Sequencing 3 Metagenomic Sequencing |
| 6 | Pig Small Intestine Tissue | Diarrhea in piglets | PCR 1: PEDV, TGEV, PDCoV, PoRV Bacterial Culture 2 + 16S rRNA Sequencing 3 Metagenomic Sequencing |
| 7 | Pig Lung Tissue | Sudden death in piglets | PCR 1: CSFV, ASFV, PRRSV, PRV Bacterial Culture 2 + 16S rRNA Sequencing 3 Metagenomic Sequencing |
| 8 | Cat Nasopharyngeal Swab | Respiratory symptoms | PCR 1: FCV, FHV-1, Chlamydophila felis, Mycoplasma felis, Bordetella bronchiseptica Metagenomic Sequencing |
| 9 | Cat Ascites | Abdominal distention, ascites | PCR 1: FCoV Metagenomic Sequencing |
| Sample No. | PCR Results | Bacterial Culture and Identification | Metagenomic Sequencing | Pathogenicity |
|---|---|---|---|---|
| 1 | FPV(−), FHV-1(−), FCV(−), FCoV(−), FIV(−), FeLV(−) | Not subjected to bacterial culture | RD114 retrovirus (80,572 reads; 45.3%) | The pathogenicity of the RD114 retrovirus is unknown; its relevance to leukopenia is uncertain |
| 2 | Mycoplasma felis (−), Chlamydia felis (−), FPV(+), FCV(−), FHV-1(−) | Not subjected to bacterial culture | FPV (22,210 reads; 12.9%) | FPV can cause fever and diarrhea in cats |
| 3 | CSFV(−), PRRSV(−), PCV2(−), PRV(−), ASFV(−) | Pasteurella multocida | Pasteurella multocida (11,520 reads; 1.8%) Clostridium novyi (1313 reads; 0.2%) | Pasteurella multocida can cause respiratory symptoms, and Clostridium novyi can cause sudden death in sows |
| 4 | FCV(−), FHV-1(−), Mycoplasma felis (+), Chlamydia felis (+), Bordetella bronchiseptica(−) | Not subjected to bacterial culture | Mycoplasma felis (14,404 reads; 18.7%) Chlamydia felis (5817 reads; 7.6%) | Mycoplasma felis and Chlamydia felis can cause respiratory symptoms in cats |
| 5 | CSFV(−), PRRSV(+), PRV(−), PCV2(+) | Pasteurella multocida, Streptococcus suis | PCV2 (47,944 reads; 10.9%) PRRSV (1934 reads; 0.4%) PPV (992 reads; 0.2%) Mycoplasma hyorhinis (21,555 reads; 4.9%) Pasteurella multocida (8857 reads; 2.0%) Glaesserella parasuis (3142 reads; 0.7%) Streptococcus suis (559 reads; 0.1%) | PCV2 can cause immunosuppression and slow growth. PRRSV can cause significant respiratory symptoms and death. PPV can cause reproductive disorders. Mycoplasma hyorhinis, Pasteurella multocida, Glaesserella parasuis, and Streptococcus suis can all cause respiratory symptoms. |
| 6 | PEDV(−), TGEV(−), PDCoV(−), PoRV(−) | Escherichia coli, but toxin gene test results were negative 2 | PSaV (19,359 reads; 4.9%) Escherichia coli (5758 reads; 1.5%) | PSav can cause diarrhea in piglets |
| 7 | CSFV(−), ASFV(−), PRRSV(−), PRV(−) | No pathogenic bacteria grew | GETV (29,244 reads; 27.5%) | GETV can cause neurological symptoms and death in piglets |
| 8 | FCV(−), FHV-1(+), Mycoplasma felis (−), Chlamydia felis (−), Bordetella bronchiseptica(−) | Not subjected to bacterial culture | FHV-1 (165,200 reads; 47.2%) | FHV-1 can cause respiratory symptoms in cats |
| 9 | FCoV(+) | Not subjected to bacterial culture | FCoV (30,102 reads; 3.3%) | FCoV can cause ascites in cats |
| Sample No. | Host Removal | Host Reads (%) | PCV2 Reads (%) | PPV Reads (%) | PRRSV Reads (%) | FHV Reads (%) | FCoV Reads (%) | Klebsiella pneumoniae Reads (%) | Mycoplasma hyorhinis Reads (%) | Streptococcus suis Reads (%) | Pasteurella multocida Reads (%) | Glaesserella parasuis Reads (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | No | 77 | 1.10 | 0.010 | 0.00086 | - | - | 0.52 | 1.45 | 0.0095 | 6.24 | 1.73 |
| Yes | 67 | 10.86 | 0.26 | 0.55 | - | - | 0.55 | 4.88 | 0.13 | 2.01 | 0.71 | |
| 8 | No | 98 | - | - | - | 0.4 | - | - | - | - | - | - |
| Yes | 48 | - | - | - | 47 | - | - | - | - | - | - | |
| 9 | No | 82 | - | - | - | - | 1.6 | - | - | - | - | - |
| Yes | 73 | - | - | - | - | 3.3 | - | - | - | - | - |
| Sample No. | Virus | Genome Type | Reference Strain NCBI ID | Reference Genome Size (bp) | Data Volume for >90% Completion 1 | Completeness for >90% Completion 2 | Viral Reads for >90% Completion /Reference Genome Size 3 | Total Data Volume 1 | Completeness for Total Data 2 | Viral Reads /Reference Genome Size 3 | Variant Bases 4 (bp) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | FPV | Linear ssDNA | MT614366.1 | 5125 | 7.3 MB | 92.9% | 0.13X | 240.5 MB | 95.4% | 4.3X | 0 |
| 5 | PCV2 | Circular ssDNA | NC006232.1 | 1767 | 3.3 MB | 100% | 0.15X | 589.4 MB | 100% | 27X | 0 |
| 6 | PSaV | Linear ssRNA | JX678943.1 | 7342 | 7.8 MB | 91.9% | 0.03X | 630.8 MB | Near 100% | 2.6X | 0 |
| 7 | GETV | Linear ssRNA | NC006558.1 | 11,597 | 6.5 MB | 98.1% | 0.13X | 125.6 MB | 99.6% | 2.5X | 2 |
| 8 | FHV-1 | Linear dsDNA | NC013590.2 | 135,797 | 98.3 MB | 93.8% | 0.2X | 594.1 MB | 98.8% | 1.2X | 0 |
| 9 | FCoV | Linear ssRNA | MW030108.1 | 29,234 | 37.1 MB | 95.3% | 0.025X | 1.6 GB | 97.5% | 1X | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, X.; Xia, Z. Application of Host-Depleted Nanopore Metagenomic Sequencing in the Clinical Detection of Pathogens in Pigs and Cats. Animals 2023, 13, 3838. https://doi.org/10.3390/ani13243838
Han X, Xia Z. Application of Host-Depleted Nanopore Metagenomic Sequencing in the Clinical Detection of Pathogens in Pigs and Cats. Animals. 2023; 13(24):3838. https://doi.org/10.3390/ani13243838
Chicago/Turabian StyleHan, Xu, and Zhaofei Xia. 2023. "Application of Host-Depleted Nanopore Metagenomic Sequencing in the Clinical Detection of Pathogens in Pigs and Cats" Animals 13, no. 24: 3838. https://doi.org/10.3390/ani13243838
APA StyleHan, X., & Xia, Z. (2023). Application of Host-Depleted Nanopore Metagenomic Sequencing in the Clinical Detection of Pathogens in Pigs and Cats. Animals, 13(24), 3838. https://doi.org/10.3390/ani13243838