
One Health and Cattle Genetic Resources: Mining More than 500 Cattle Genomes to Identify Variants in Candidate Genes Potentially Affecting Coronavirus Infections



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Re-Sequencing Datasets
2.2. Sequence Alignment and Variant Detection
2.3. Comparative Analysis between Cattle and Human Protein Sequences of Candidate Genes
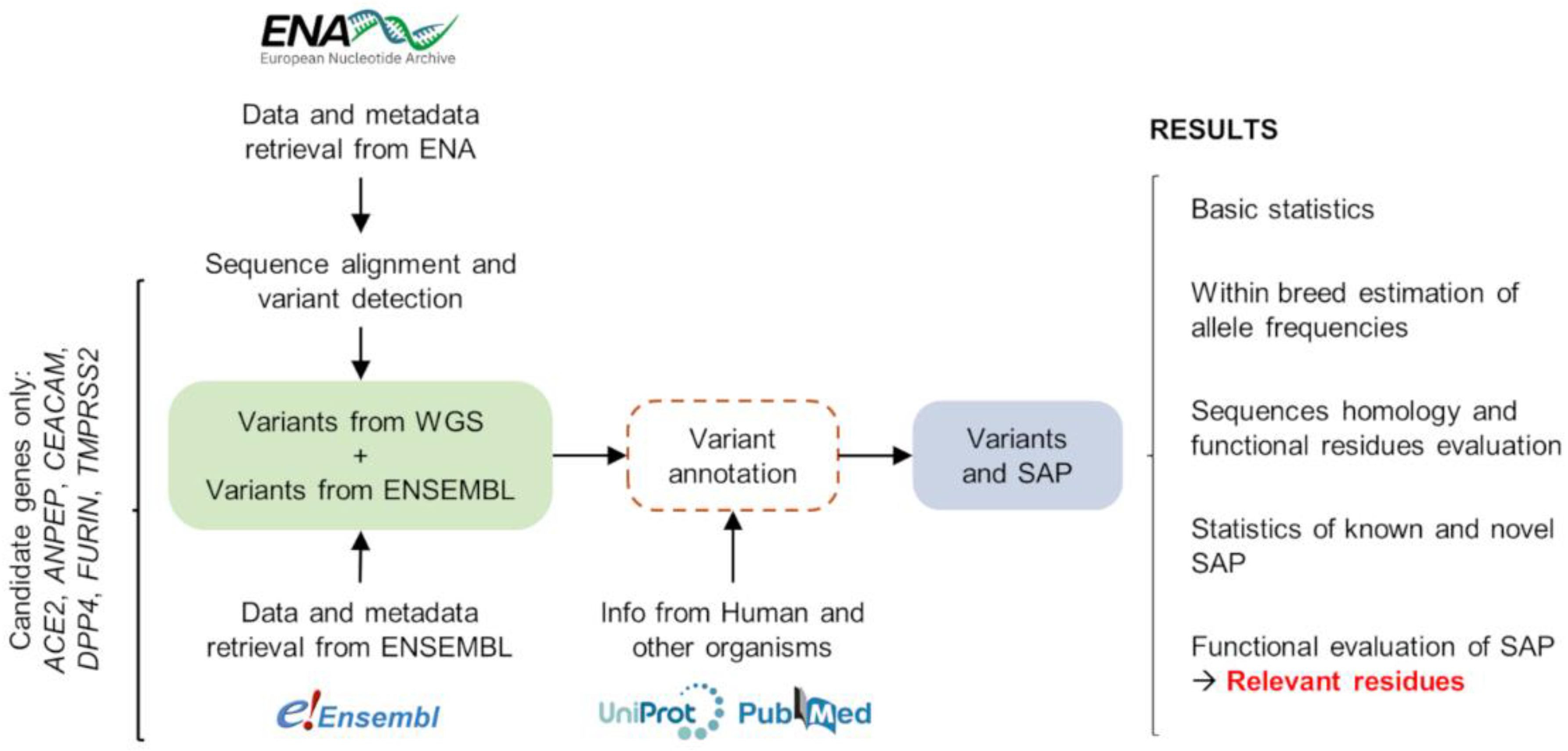
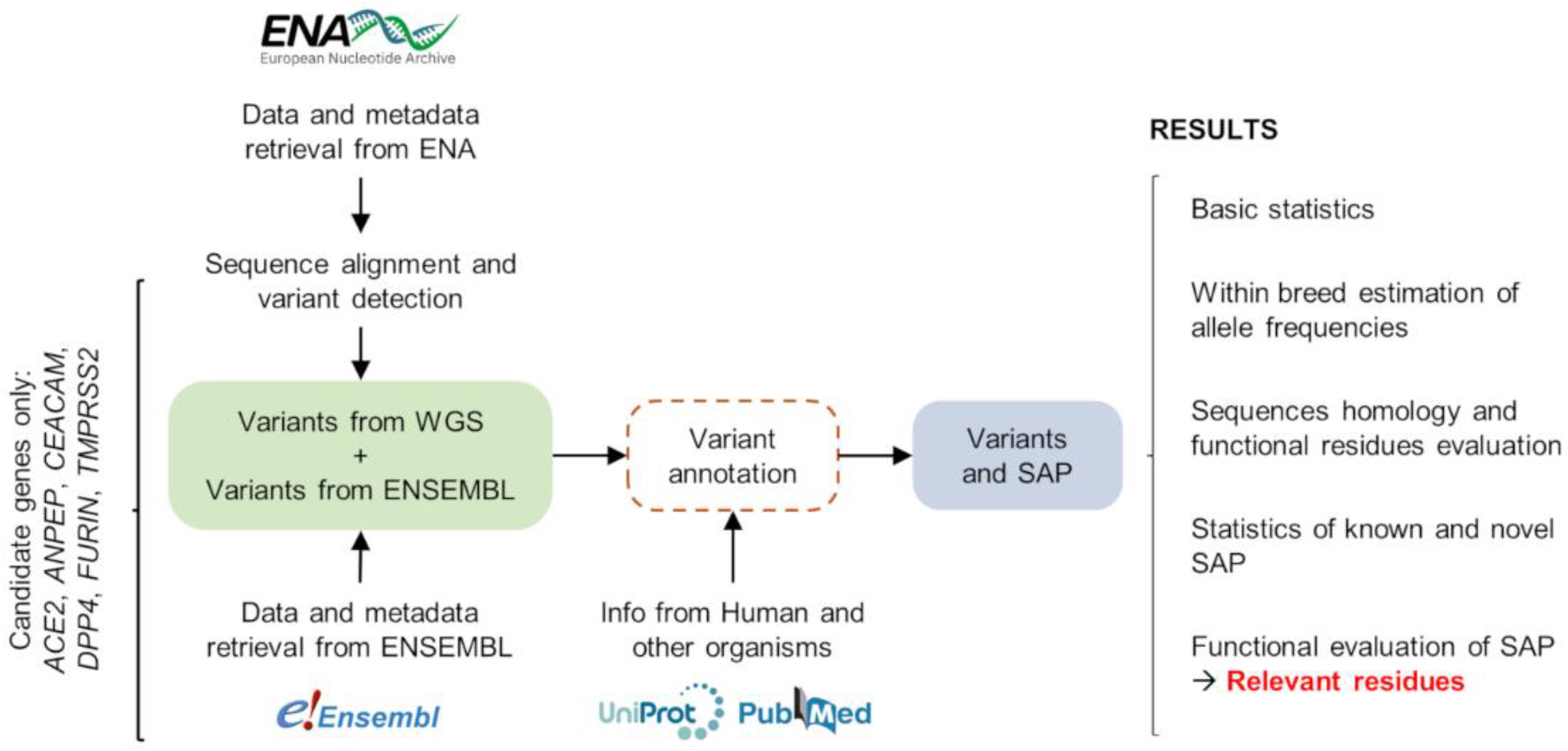
2.4. Overview of the Bioinformatic Pipelines
3. Results
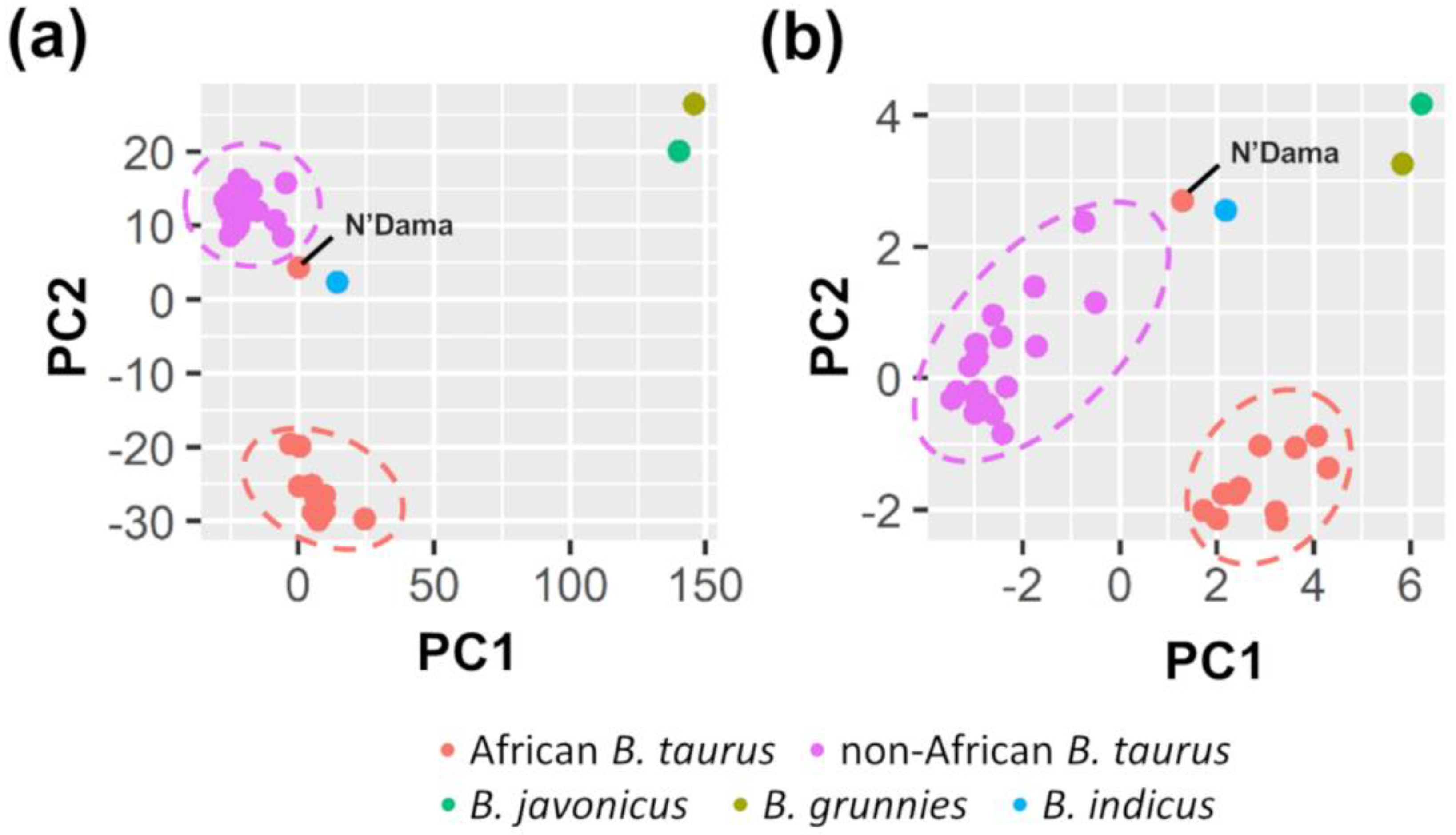
3.1. Sequencing Results and Variability Detected in Candidate Genes
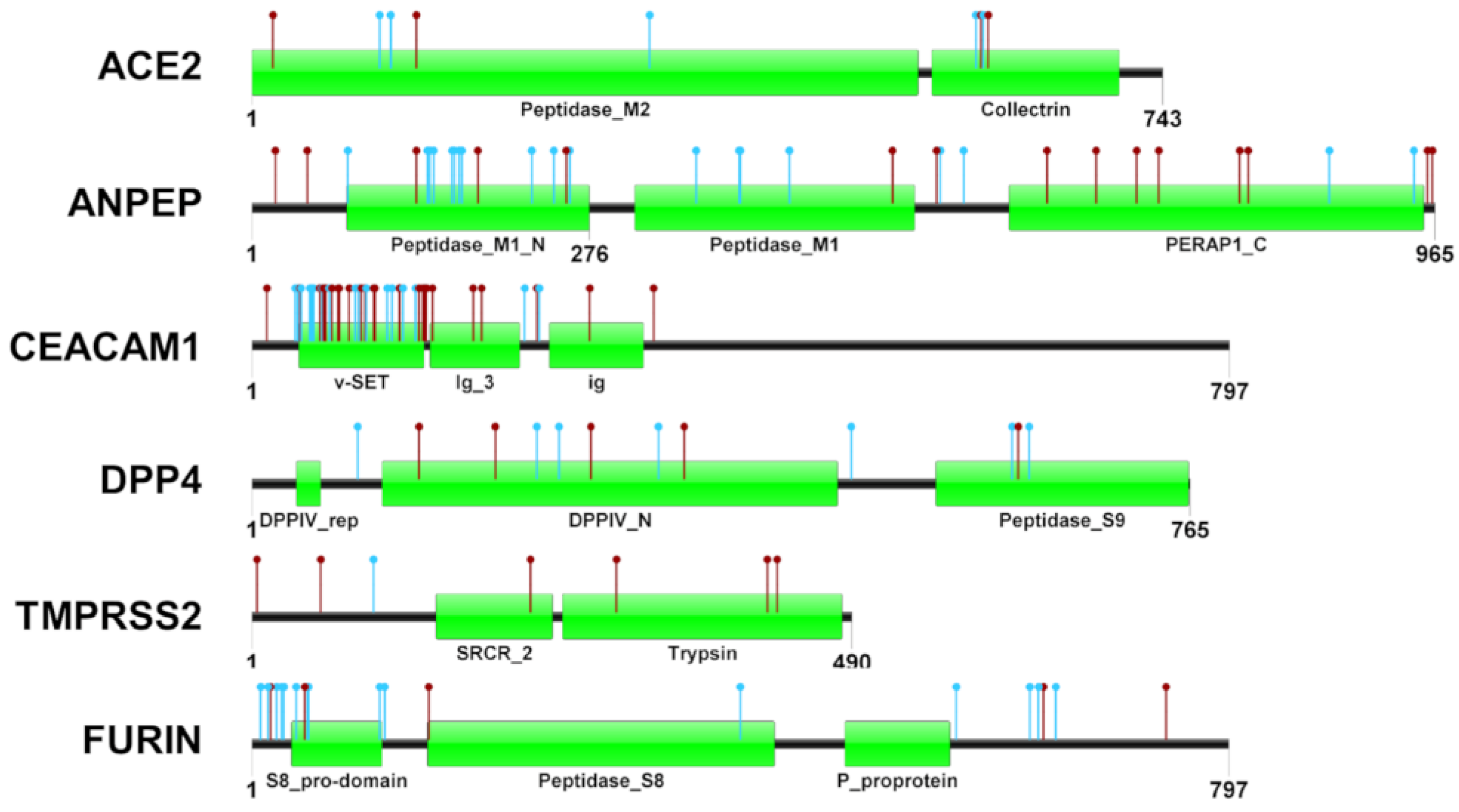
3.2. Protein Variants Affecting Candidate Genes
3.3. Functional Evaluation of Protein Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Veterinary Medical Association. One Health: A New Professional Imperative. One Health Initiative Task Force: Final Report. 2008. Available online: https://www.avma.org/sites/default/files/resources/onehealth_final.pdf (accessed on 7 January 2022).
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global Trends in Emerging Infectious Diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Munster, V.J.; Koopmans, M.; van Doremalen, N.; van Riel, D.; de Wit, E. A Novel Coronavirus Emerging in China-Key Questions for Impact Assessment. N. Engl. J. Med. 2020, 382, 692–694. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A Novel Coronavirus Outbreak of Global Health Concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Harapan, H.; Itoh, N.; Yufika, A.; Winardi, W.; Keam, S.; Te, H.; Megawati, D.; Hayati, Z.; Wagner, A.L.; Mudatsir, M. Coronavirus Disease 2019 (COVID-19): A Literature Review. J. Infect. Public Health 2020, 13, 667–673. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the Origin and Continuing Evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020, 30, 1346–1351.e2. [Google Scholar] [CrossRef]
- Haake, C.; Cook, S.; Pusterla, N.; Murphy, B. Coronavirus Infections in Companion Animals: Virology, Epidemiology, Clinical and Pathologic Features. Viruses 2020, 12, 1023. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Lorusso, A. Novel Human Coronavirus (SARS-CoV-2): A Lesson from Animal Coronaviruses. Vet. Microbiol. 2020, 244, 108693. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of Ferrets, Cats, Dogs, and Other Domesticated Animals to SARS-Coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef] [Green Version]
- Cavanagh, D. Coronaviruses in Poultry and Other Birds. Avian Pathol. 2005, 34, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Rabozzi, G. Emerging Zoonoses: A One Health Challenge. EClinicalMedicine 2020, 19, 100300. [Google Scholar] [CrossRef]
- Groat, J.D. Lessons of Past Coronavirus Pandemics. Popul. Dev. Rev. 2020, 46, 633–637. [Google Scholar] [CrossRef]
- Hodnik, J.J.; Ježek, J.; Starič, J. Coronaviruses in Cattle. Trop. Anim. Health. Prod. 2020, 52, 2809–2816. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Saif, L.J. Bovine Coronavirus and the Associated Diseases. Front. Vet. Sci. 2021, 8, 643220. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Drigo, M.; Legnardi, M.; Grassi, L.; Pasotto, D.; Menandro, M.L.; Cecchinato, M.; Tucciarone, C.M. Bovine Coronavirus: Variability, Evolution, and Dispersal Patterns of a No Longer Neglected Betacoronavirus. Viruses 2020, 12, 1285. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Peng, G.; Sun, D.; Rajashankar, K.R.; Qian, Z.; Holmes, K.V.; Li, F. Crystal Structure of Mouse Coronavirus Receptor-Binding Domain Complexed with Its Murine Receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 10696–10701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 Is a Host Factor for SARS-CoV-2 Infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Kyrou, I.; Randeva, H.S.; Spandidos, D.A.; Karteris, E. Not Only ACE2—the Quest for Additional Host Cell Mediators of SARS-CoV-2 Infection: Neuropilin-1 (NRP1) as a Novel SARS-CoV-2 Host Cell Entry Mediator Implicated in COVID-19. Sig. Transduct. Target Ther. 2021, 6, 21. [Google Scholar] [CrossRef]
- Asselta, R.; Paraboschi, E.M.; Mantovani, A.; Duga, S. ACE2 and TMPRSS2 Variants and Expression as Candidates to Sex and Country Differences in COVID-19 Severity in Italy. Aging 2020, 12, 10087–10098. [Google Scholar] [CrossRef]
- Benetti, E.; Tita, R.; Spiga, O.; Ciolfi, A.; Birolo, G.; Bruselles, A.; Doddato, G.; Giliberti, A.; Marconi, C.; Musacchia, F.; et al. ACE2 Gene Variants May Underlie Interindividual Variability and Susceptibility to COVID-19 in the Italian Population. Eur. J. Hum. Genet. 2020, 28, 1602–1614. [Google Scholar] [CrossRef]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative Genetic Analysis of the Novel Coronavirus (2019-NCoV/SARS-CoV-2) Receptor ACE2 in Different Populations. Cell Discov. 2020, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.A.; Amanullah, A.; Aziz, B. Structural Variations in Human ACE2 May Influence Its Binding with SARS-CoV-2 Spike Protein. J. Med. Virol. 2020, 92, 1580–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryamohan, K.; Diwanji, D.; Stawiski, E.W.; Gupta, R.; Miersch, S.; Liu, J.; Chen, C.; Jiang, Y.-P.; Fellouse, F.A.; Sathirapongsasuti, J.F.; et al. Human ACE2 Receptor Polymorphisms and Altered Susceptibility to SARS-CoV-2. Commun. Biol. 2021, 4, 475. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Schiavo, G.; Ribani, A.; Utzeri, V.J.; Taurisano, V.; Ballan, M.; Muñoz, M.; Alves, E.; Araujo, J.P.; Bozzi, R.; et al. Describing Variability in Pig Genes Involved in Coronavirus Infections for a One Health Perspective in Conservation of Animal Genetic Resources. Sci. Rep. 2021, 11, 3359. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.W.; Ahamed, A.; Aslam, R.; Alako, B.T.F.; Burgin, J.; Buso, N.; Courtot, M.; Fan, J.; Gupta, D.; Haseeb, M.; et al. The European Nucleotide Archive in 2020. Nucleic Acids Res. 2020, 49, D82–D85. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; der Auwera, G.A.V.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. bioRxiv 2018, 201178. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Raskin, L.; Samuels, D.C.; Shyr, Y.; Guo, Y. Genome Measures Used for Quality Control Are Dependent on Gene Function and Ancestry. Bioinformatics 2015, 31, 318–323. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Sievers, F.; Higgins, D.G. Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. Methods Mol. Biol. 2014, 1079, 105–116. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kwon, T.; Dessie, T.; Yoo, D.; Mwai, O.A.; Jang, J.; Sung, S.; Lee, S.; Salim, B.; Jung, J.; et al. The Mosaic Genome of Indigenous African Cattle as a Unique Genetic Resource for African Pastoralism. Nat. Genet. 2020, 52, 1099–1110. [Google Scholar] [CrossRef]
- Reguera, J.; Santiago, C.; Mudgal, G.; Ordoño, D.; Enjuanes, L.; Casasnovas, J.M. Structural Bases of Coronavirus Attachment to Host Aminopeptidase N and Its Inhibition by Neutralizing Antibodies. PLoS Pathog. 2012, 8, e1002859. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.; Murray, K.A.; Zambrana-Torrelio, C.; Morse, S.S.; Rondinini, C.; Di Marco, M.; Breit, N.; Olival, K.J.; Daszak, P. Global Hotspots and Correlates of Emerging Zoonotic Diseases. Nat. Commun. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Vincent, A.L. Reverse Zoonosis of Influenza to Swine: New Perspectives on the Human-Animal Interface. Trends Microbiol. 2015, 23, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Bishop, S.C.; Axford, R.F.E.; Nicholas, F.W.; Owen, J.B. Breeding for Disease Resistance in Farm Animals, 3rd ed.; CABI: Wallingford, UK, 2010; ISBN 978-1-84593-555-9. [Google Scholar]
- Neibergs, H.L.; Seabury, C.M.; Wojtowicz, A.J.; Wang, Z.; Scraggs, E.; Kiser, J.N.; Neupane, M.; Womack, J.E.; Van Eenennaam, A.; Hagevoort, G.R.; et al. Susceptibility Loci Revealed for Bovine Respiratory Disease Complex in Pre-Weaned Holstein Calves. BMC Genom. 2014, 15, 1164. [Google Scholar] [CrossRef] [Green Version]
- Kiser, J.N.; Lawrence, T.E.; Neupane, M.; Seabury, C.M.; Taylor, J.F.; Womack, J.E.; Neibergs, H.L. Rapid Communication: Subclinical Bovine Respiratory Disease-Loci and Pathogens Associated with Lung Lesions in Feedlot Cattle. J. Anim. Sci. 2017, 95, 2726–2731. [Google Scholar] [CrossRef]
- Kiser, J.N.; Neibergs, H.L. Identifying Loci Associated With Bovine Corona Virus Infection and Bovine Respiratory Disease in Dairy and Feedlot Cattle. Front. Vet. Sci. 2021, 8, 679074. [Google Scholar] [CrossRef]
- Neibergs, H.; Zanella, R.; Casas, E.; Snowder, G.D.; Wenz, J.; Neibergs, J.S.; Moore, D. Loci on Bos Taurus Chromosome 2 and Bos Taurus Chromosome 26 Are Linked with Bovine Respiratory Disease and Associated with Persistent Infection of Bovine Viral Diarrhea Virus. J. Anim. Sci. 2011, 89, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavassoly, I.; Goldfarb, J.; Iyengar, R. Systems Biology Primer: The Basic Methods and Approaches. Essays Biochem. 2018, 62, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Damas, J.; Hughes, G.M.; Keough, K.C.; Painter, C.A.; Persky, N.S.; Corbo, M.; Hiller, M.; Koepfli, K.-P.; Pfenning, A.R.; Zhao, H.; et al. Broad Host Range of SARS-CoV-2 Predicted by Comparative and Structural Analysis of ACE2 in Vertebrates. Proc. Natl. Acad. Sci. USA 2020, 117, 22311–22322. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Jin, X.; Lu, Y.; Zhang, L. SARS-CoV-2 Spike Protein Favors ACE2 from Bovidae and Cricetidae. J. Med. Virol. 2020, 92, 1649–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Zhao, Y.-B.; Wang, Q.; Li, J.-Y.; Zhou, Z.-J.; Liao, C.-H.; Ge, X.-Y. Predicting the Angiotensin Converting Enzyme 2 (ACE2) Utilizing Capability as the Receptor of SARS-CoV-2. Microbes Infect. 2020, 22, 221–225. [Google Scholar] [CrossRef]
- Low-Gan, J.; Huang, R.; Kelley, A.; Jenkins, G.W.; McGregor, D.; Smider, V.V. Diversity of ACE2 and its interaction with SARS-CoV-2 receptor binding domain. Biochem. J. 2021, 478, 3671–3684. [Google Scholar] [CrossRef]
- Alexander, M.R.; Schoeder, C.T.; Brown, J.A.; Smart, C.D.; Moth, C.; Wikswo, J.P.; Capra, J.A.; Meiler, J.; Chen, W.; Madhur, M.S. Predicting Susceptibility to SARS-CoV-2 Infection Based on Structural Differences in ACE2 across Species. FASEB J. 2020, 34, 15946–15960. [Google Scholar] [CrossRef]
- Ulrich, L.; Wernike, K.; Hoffmann, D.; Mettenleiter, T.C.; Beer, M. Experimental Infection of Cattle with SARS-CoV-2. Emerg. Infect. Dis. 2020, 26, 2979–2981. [Google Scholar] [CrossRef]
- Falkenberg, S.; Buckley, A.; Laverack, M.; Martins, M.; Palmer, M.V.; Lager, K.; Diel, D.G. Experimental Inoculation of Young Calves with SARS-CoV-2. Viruses 2021, 13, 441. [Google Scholar] [CrossRef]
- Mick, E.; Kamm, J.; Pisco, A.O.; Ratnasiri, K.; Babik, J.M.; Castañeda, G.; DeRisi, J.L.; Detweiler, A.M.; Hao, S.L.; Kangelaris, K.N.; et al. Upper Airway Gene Expression Reveals Suppressed Immune Responses to SARS-CoV-2 Compared with Other Respiratory Viruses. Nat. Commun. 2020, 11, 5854. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; He, W.-T.; Wang, L.; Lai, A.; Ji, X.; Zhai, X.; Li, G.; Suchard, M.A.; Tian, J.; Zhou, J.; et al. COVID-19: Epidemiology, evolution, and cross-disciplinary perspectives. Trends Mol. Med. 2020, 26, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.-K.; Huang, I.-C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, T.Z.; Bunkenborg, J.; Gronborg, M.; Molina, H.; Thuluvath, P.J.; Argani, P.; Goggins, M.G.; Maitra, A.; Pandey, A. A proteomic analysis of human bile. Mol. Cell. Proteom. 2004, 3, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Jiang, X.; Sun, D.; Han, G.; Wang, F.; Ye, M.; Wang, L.; Zou, H. Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J. Proteome Res. 2009, 8, 651–661. [Google Scholar] [CrossRef]
- Li, Z.; Tomlinson, A.C.; Wong, A.H.; Zhou, D.; Desforges, M.; Talbot, P.J.; Benlekbir, S.; Rubinstein, J.L.; Rini, J.M. The human coronavirus HCoV-229E S-protein structure and receptor binding. Elife 2019, 8, e51230. [Google Scholar] [CrossRef]
- Wentworth, D.E.; Holmes, K.V. Molecular determinants of species specificity in the coronavirus receptor aminopeptidase N (CD13): Influence of N-linked glycosylation. J. Virol. 2001, 75, 9741–9752. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.H.; Zhou, D.; Rini, J.M. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J. Biol. Chem. 2012, 287, 36804–36813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, A.F.; Maile, J.; Heister, A.; Siddell, S.G. Characterization of functional domains in the human coronavirus HCV 229E receptor. J. Gen. Virol. 1996, 77, 2515–2521. [Google Scholar] [CrossRef] [PubMed]
- Delmas, B.; Gelfi, J.; Kut, E.; Sjöström, H.; Noren, O.; Laude, H. Determinants essential for the transmissible gastroenteritis virus-receptor interaction reside within a domain of aminopeptidase-N that is distinct from the enzymatic site. J. Virol. 1994, 68, 5216–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundry, R.L.; Raginski, K.; Tarasova, Y.; Tchernyshyov, I.; Bausch-Fluck, D.; Elliott, S.T.; Boheler, K.R.; Van Eyk, J.E.; Wollscheid, B. The mouse C2C12 myoblast cell surface N-linked glycoproteome: Identification, glycosite occupancy, and membrane orientation. Mol. Cell. Proteom. 2009, 8, 2555–2569. [Google Scholar] [CrossRef] [Green Version]
- Wollscheid, B.; Bausch-Fluck, D.; Henderson, C.; O’Brien, R.; Bibel, M.; Schiess, R.; Aebersold, R.; Watts, J.D. Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nat. Biotechnol. 2009, 27, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Guo, D.; Fu, L.; Cui, Y.; Liu, X.; et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef] [Green Version]
- de Wit, E.; Feldmann, F.; Horne, E.; Martellaro, C.; Haddock, E.; Bushmaker, T.; Rosenke, K.; Okumura, A.; Rosenke, R.; Saturday, G.; et al. Domestic pig unlikely reservoir for MERS-CoV. Emerg. Infect. Dis. 2017, 23, 985–988. [Google Scholar] [CrossRef] [Green Version]
- Kirby, M.; Yu, D.M.; O’Connor, S.; Gorrell, M.D. Inhibitor selectivity in the clinical application of dipeptidyl peptidase-4 inhibition. Clin. Sci. 2010, 118, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Hiramatsu, H.; Kyono, K.; Higashiyama, Y.; Fukushima, C.; Shima, H.; Sugiyama, S.; Inaka, K.; Yamamoto, A.; Shimizu, R. The structure and function of human dipeptidyl peptidase IV, possessing a unique eight-bladed β-propeller fold. Biochem. Biophys. Res. Commun. 2003, 302, 849–854. [Google Scholar] [CrossRef]
- Rasmussen, H.B.; Branner, S.; Wiberg, F.C.; Wagtmann, N. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nat. Struct. Biol. 2003, 10, 19–25. [Google Scholar] [CrossRef]
- Thoma, R.; Löffler, B.; Stihle, M.; Huber, W.; Ruf, A. Hennig M. Structural basis of proline-specific exopeptidase activity as observed in human dipeptidyl peptidase-IV. Structure 2003, 11, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Meng, W.; Brigance, R.P.; Chao, H.J.; Fura, A.; Harrity, T.; Marcinkeviciene, J.; O’Connor, S.P.; Tamura, J.K.; Xie, D.; Zhang, Y.; et al. Discovery of 6-(Aminomethyl)-5-(2, 4-dichlorophenyl)-7-methylimidazo [1, 2-a] pyrimidine-2-carboxamides as Potent, Selective Dipeptidyl Peptidase-4 (DPP4) Inhibitors. J. Med. Chem. 2010, 53, 5620–5628. [Google Scholar] [CrossRef] [PubMed]
- Afar, D.E.; Vivanco, I.; Hubert, R.S.; Kuo, J.; Chen, E.; Saffran, D.C.; Raitano, A.B.; Jakobovits, A. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 2001, 61, 1686–1692. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Ensembl Transcript | UniprotKB ACC | N. of Variants from WGS 1 | N. of SAP from WGS 2 | N. of SAP from Ensembl |
|---|---|---|---|---|---|
| ACE2 | ENSBTAT00000048730.4 | A0A452DJE0 | 1251 (358) | 15 (4 + 6 + 5) | 155 |
| ANPEP | ENSBTAT00000068383.1 | A0A3Q1MB09 | 464 (112) | 45 (19 + 18 + 8) | 73 |
| CEACAM1 | ENSBTAT00000069303.1 | A0A3Q1MQ27 | 723 (212) | 62 (41 + 7 + 14) | 104 |
| DPP4 | ENSBTAT00000056886.3.1 | P81425 | 2270 (716) | 18 (3 + 9 + 6) | 67 |
| FURIN | ENSBTAT00000072776.1 | B0JYR0 | 249 (71) | 30 (9 + 13 + 8) | 219 |
| TMPRSS2 | ENSBTAT00000012036.5.2 | A2VDV7 | 1608 (526) | 10 (6 + 1 + 3) | 90 |
| Total | 6565 (1995) | 180 (82 + 54 + 44) | 708 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bovo, S.; Schiavo, G.; Fontanesi, L. One Health and Cattle Genetic Resources: Mining More than 500 Cattle Genomes to Identify Variants in Candidate Genes Potentially Affecting Coronavirus Infections. Animals 2022, 12, 838. https://doi.org/10.3390/ani12070838
Bovo S, Schiavo G, Fontanesi L. One Health and Cattle Genetic Resources: Mining More than 500 Cattle Genomes to Identify Variants in Candidate Genes Potentially Affecting Coronavirus Infections. Animals. 2022; 12(7):838. https://doi.org/10.3390/ani12070838
Chicago/Turabian StyleBovo, Samuele, Giuseppina Schiavo, and Luca Fontanesi. 2022. "One Health and Cattle Genetic Resources: Mining More than 500 Cattle Genomes to Identify Variants in Candidate Genes Potentially Affecting Coronavirus Infections" Animals 12, no. 7: 838. https://doi.org/10.3390/ani12070838
APA StyleBovo, S., Schiavo, G., & Fontanesi, L. (2022). One Health and Cattle Genetic Resources: Mining More than 500 Cattle Genomes to Identify Variants in Candidate Genes Potentially Affecting Coronavirus Infections. Animals, 12(7), 838. https://doi.org/10.3390/ani12070838