
Assessing Genomic Diversity and Selective Pressures in Bohai Black Cattle Using Whole-Genome Sequencing Data

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Whole-Genome Sequencing
2.2. Reads Mapping and SNP Calling
2.3. Population Genomic Parameter Analysis
2.4. Population Genetic Structure and Phylogenetic Analysis
2.5. Genomic Signatures of Positive Selection
3. Results
3.1. Sequencing and SNPs Calling
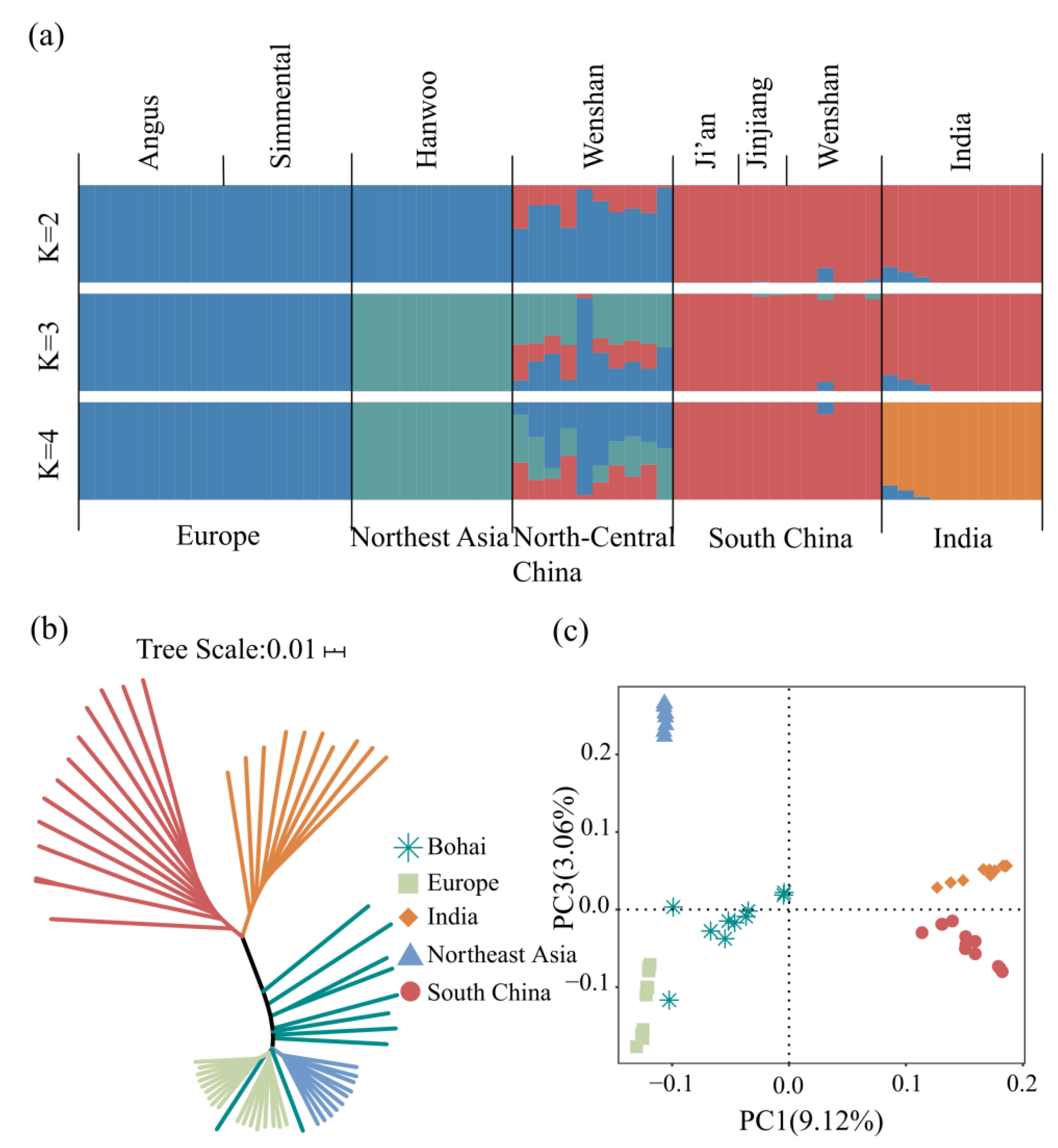
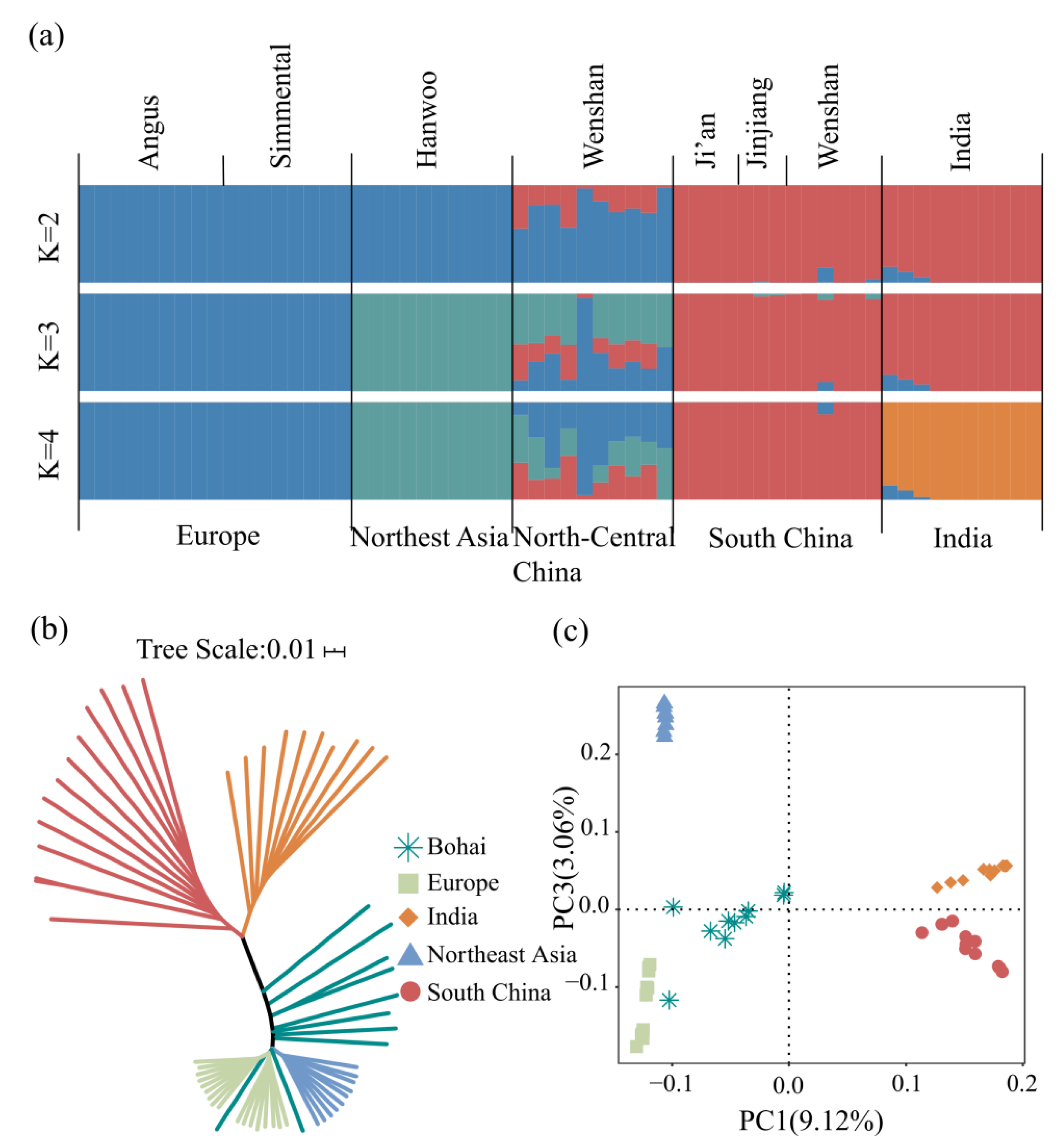
3.2. Population Genetic Structure and Phylogenetic Analysis
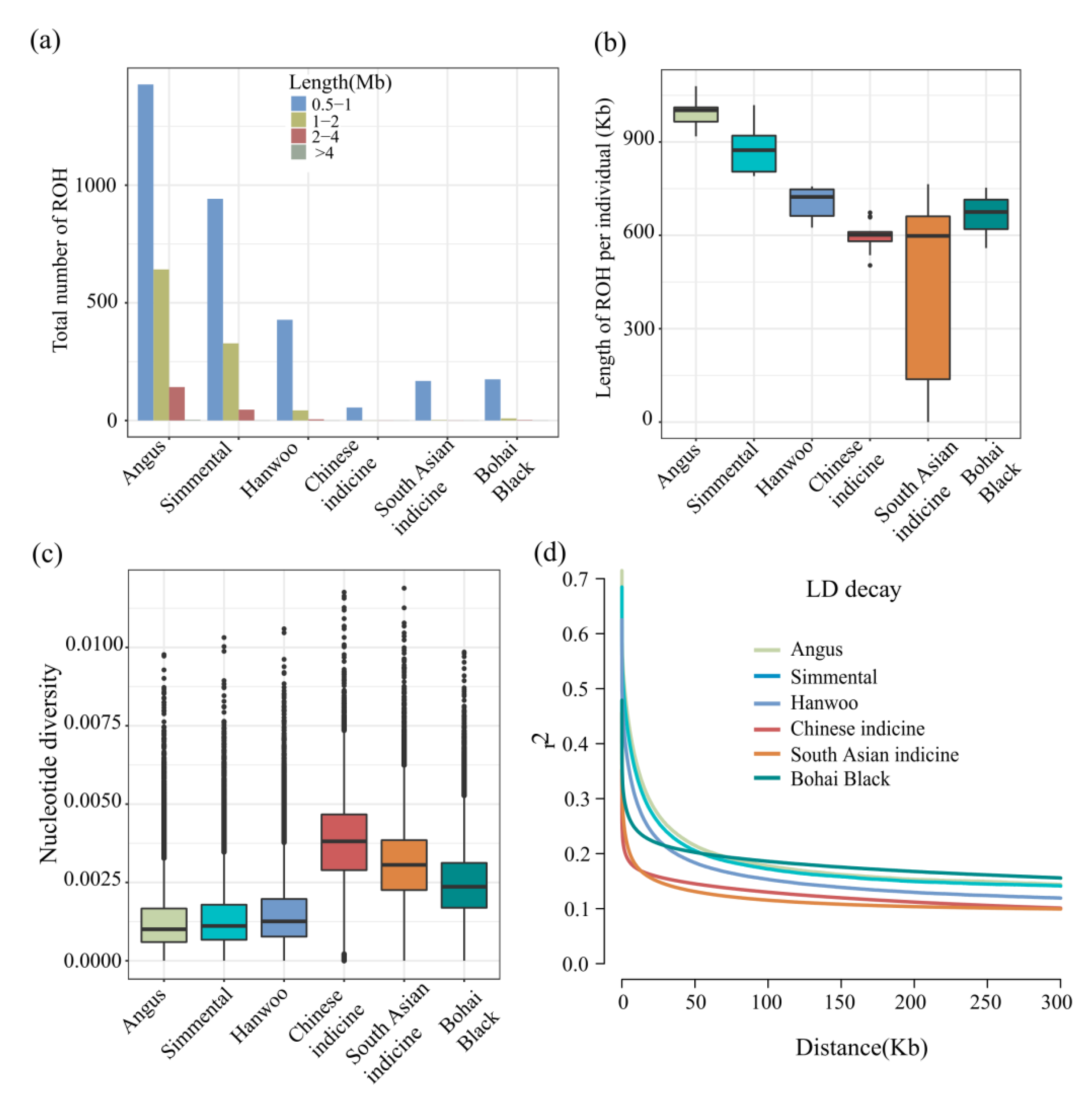
3.3. Population Genomic Parameter Analysis
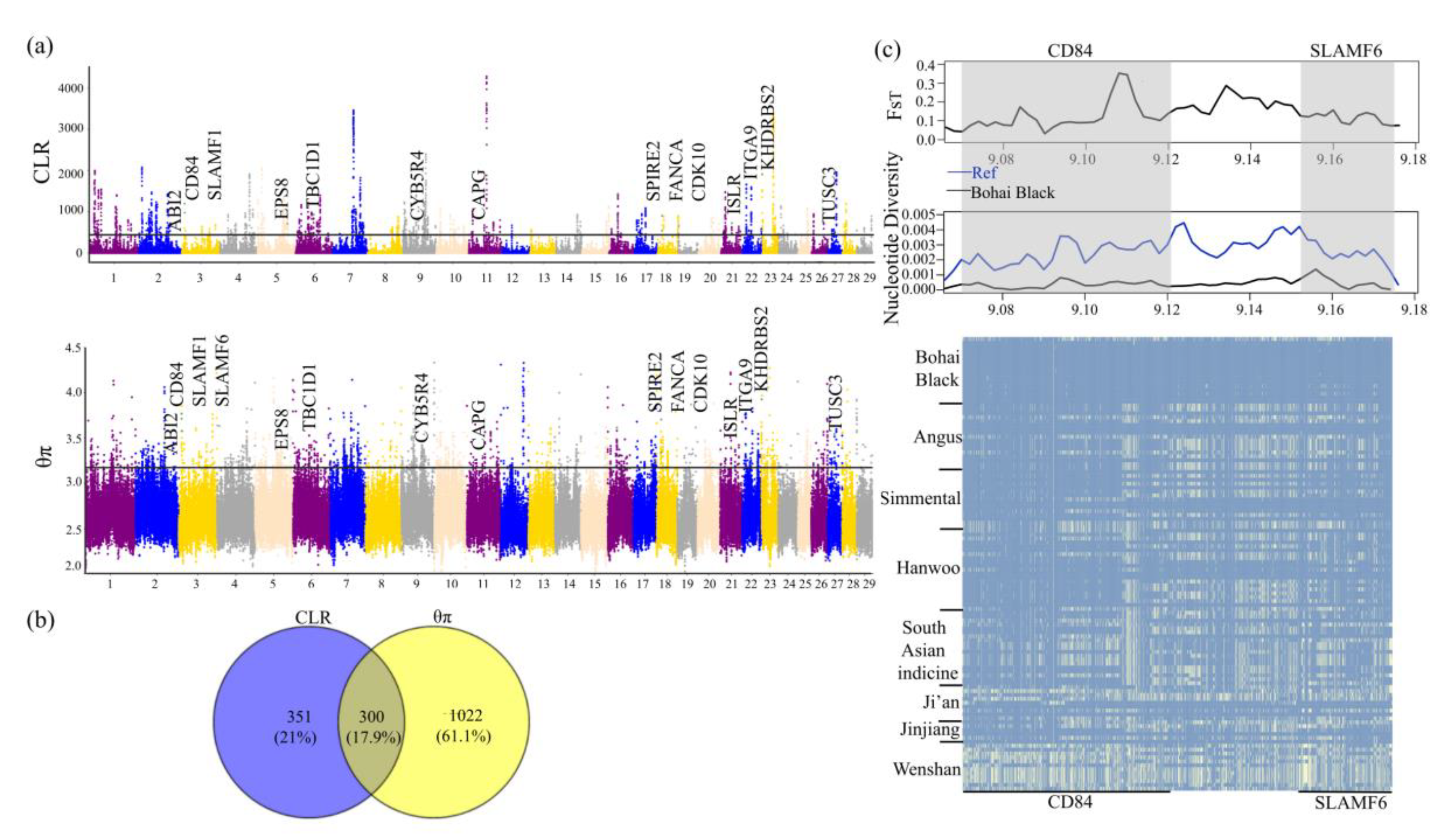
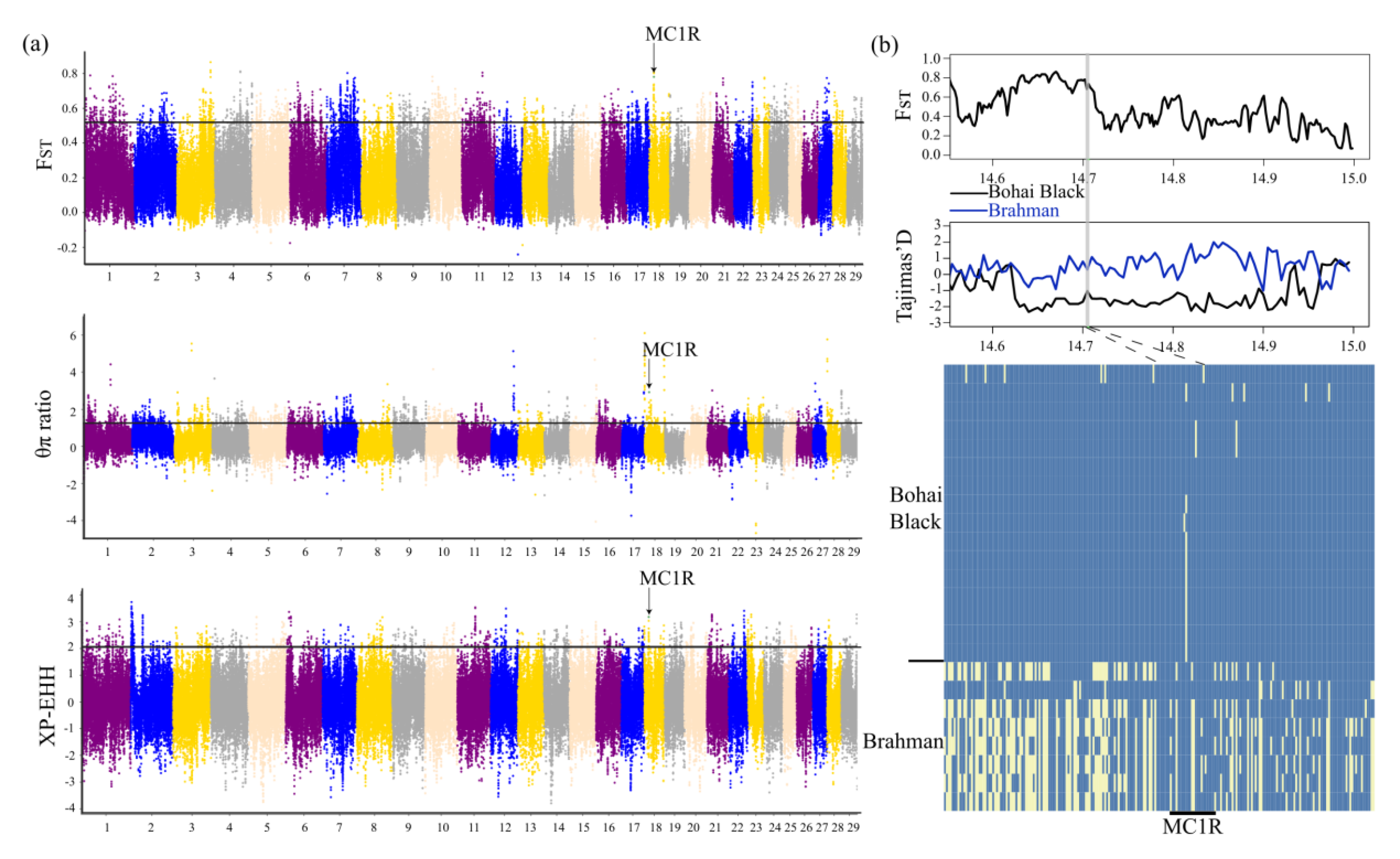
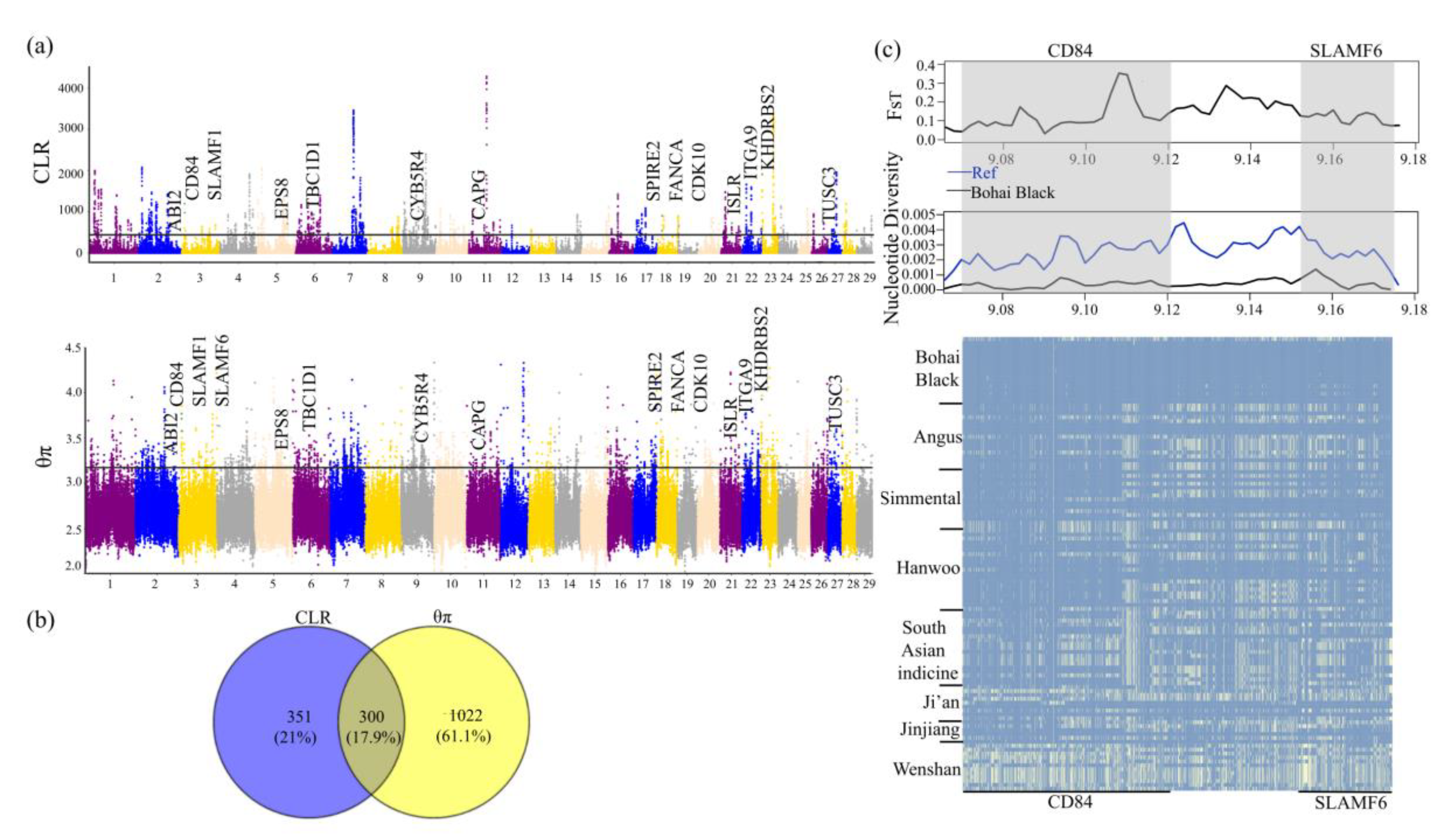
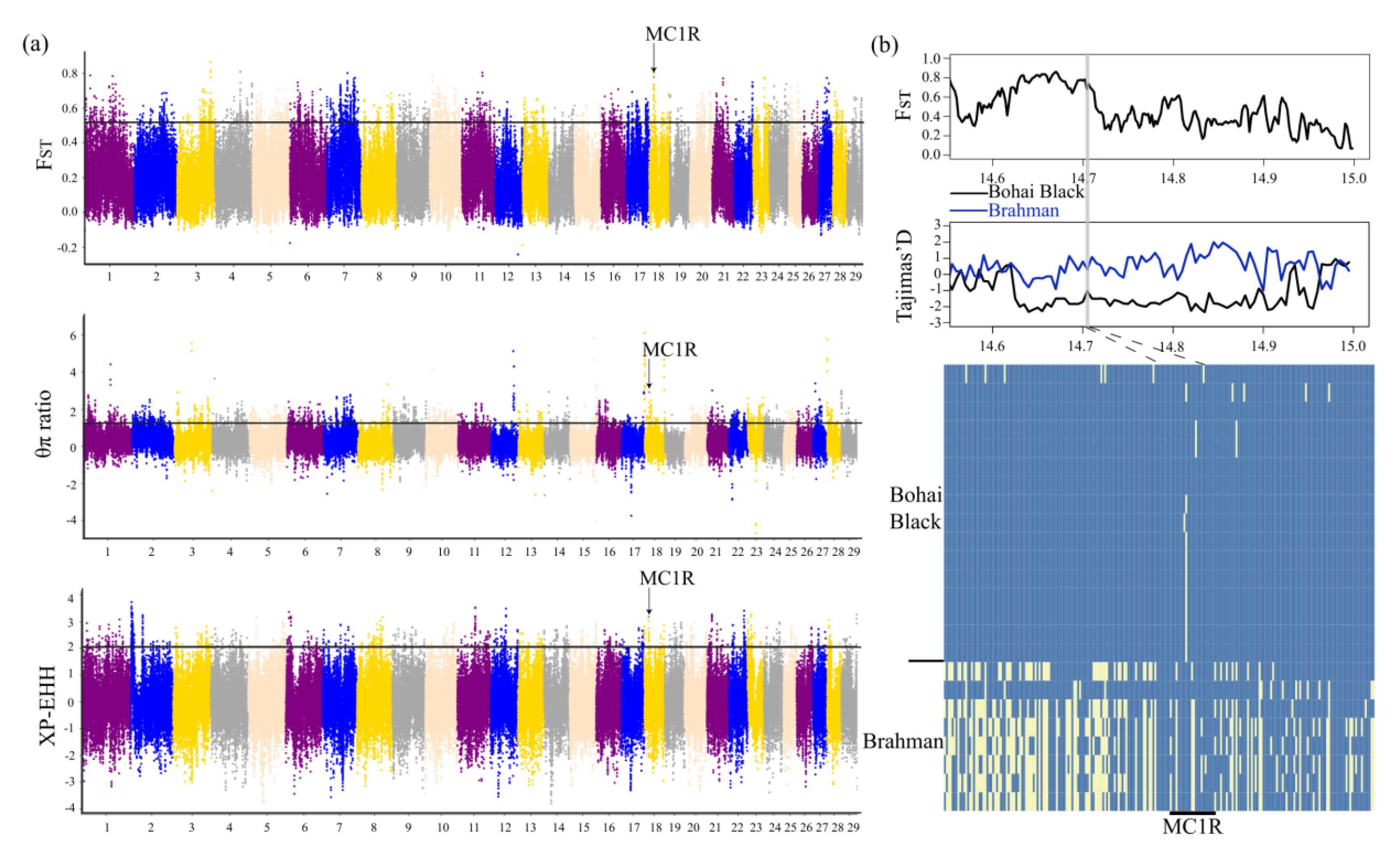
3.4. Genomic Signatures of Positive Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frantz, L.A.F.; Bradley, D.G.; Larson, G.; Orlando, L. Animal domestication in the era of ancient genomics. Nat. Rev. Genet. 2020, 21, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Lenstra, J.A.; Zhang, S.; Liu, W.; Liu, J. Evolution and domestication of the Bovini species. Anim. Genet. 2020, 51, 637–657. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-D.; Ding, X.-D.; Wang, S.; Wójcik, J.; Zhang, Y.; Tokarska, M.; Li, Y.; Wang, M.-S.; Faruque, O.; Nielsen, R.; et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat. Ecol. Evol. 2018, 2, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Hailer, F.; Upadhyay, M.; Del Corvo, M.; Colli, L.; Negrini, R.; Kim, E.-S.; Crooijmans, R.P.M.A.; Sonstegard, T.; Ajmone-Marsan, P. Adaptive introgression from indicine cattle into white cattle breeds from Central Italy. Sci. Rep. 2020, 10, 1279. [Google Scholar] [CrossRef]
- Illa, S.K.; Mukherjee, S.; Nath, S.; Mukherjee, A. Genome-Wide Scanning for Signatures of Selection Revealed the Putative Genomic Regions and Candidate Genes Controlling Milk Composition and Coat Color Traits in Sahiwal Cattle. Front. Genet. 2021, 12, 699422. [Google Scholar] [CrossRef]
- Chen, Q.; Huang, B.; Zhan, J.; Wang, J.; Qu, K.; Zhang, F.; Shen, J.; Jia, P.; Ning, Q.; Zhang, J.; et al. Whole-genome analyses identify loci and selective signals associated with body size in cattle. J. Anim. Sci. 2020, 98, skaa068. [Google Scholar] [CrossRef]
- Ahozonlin, M.; Dossa, L.H.; Dahouda, M.; Gbangboche, A.B. Morphological divergence in the West African shorthorn Lagune cattle populations from Benin. Trop. Anim. Health Prod. 2019, 52, 803–814. [Google Scholar] [CrossRef]
- Liu, Z.Y. Local improved breed: Bohai Black Cattle. Sichuan Anim. Husb. Vet. 2015, 42, 50. (In Chinese) [Google Scholar]
- Zhang, Z.Q. Development of new strains based on conservation of Bohai Black Cattle. Contemp. Anim. Husb. 2017, 18–19. Available online: https://kns.cnki.net/kcms/detail/detail.aspx?FileName=DDXM201723012&DbName=CJFQ2017 (accessed on 9 December 2021). (In Chinese).
- Liu, Z.Y. Investigation and study on development of Bohai Black cattle. China Anim. Husb. 2015, 47–48. Available online: https://kns.cnki.net/kcms/detail/detail.aspx?FileName=MYTX201517025&DbName=CJFQ2015 (accessed on 9 December 2021). (In Chinese).
- Mao, Y.; Chang, H.; Yang, Z.; Zhang, L.; Xu, M.; Sun, W.; Chang, G.; Song, G. Genetic Structure and Differentiation of Three Chinese Indigenous Cattle Populations. Biochem. Genet. 2007, 45, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.; Lan, Z.; Chen, J.; Wu, Y.; Song, E.; Liu, X.; Wan, F. Study on genetic variation of mtDNA D-Loop region in Bohai Black cattle and Japanese cattle. J. China Agric. Univ. 2009, 14, 75–82. (In Chinese) [Google Scholar]
- Chang, Z.; Hou, J.; Song, E.; Cheng, H.; Chen, H.; Lei, C. Study on Y chromosome genetic diversity and paternal origin of Black cattle in Bohai Black cattle. China Cattle Sci. 2018, 44, 41–44. (In Chinese) [Google Scholar]
- Gan, H.Y.; Li, J.B.; Wang, H.M.; Gao, Y.D.; Zhong, J.F. Relationship between the melanocortin receptor 1 (MC1R) gene and the coat color phenotype in cattle. Hereditas 2007, 29, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.P.; Gan, Q.F.; Zhang, X.H.; Li, H.D.; Hou, G.Y.; Li, J.Y.; Gao, X.; Ren, H.Y.; Chen, J.B.; Xu, S.Z. Detecting a deletion in the coding region of the bovine bone morphogenetic protein 15 gene (BMP15). J. Appl. Genet. 2009, 50, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Edea, Z.; Bhuiyan, M.S.A.; Dessie, T.; Rothschild, M.F.; Dadi, H.; Kim, K.S. Genome-wide genetic diversity, population structure and admixture analysis in African and Asian cattle breeds. Animal 2015, 9, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef]
- Stothard, P.; Choi, J.-W.; Basu, U.; Sumner-Thomson, J.M.; Meng, Y.; Liao, X.; Moore, S.S. Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genom. 2011, 12, 559. [Google Scholar] [CrossRef] [Green Version]
- Somavilla, A.L.; Sonstegard, T.S.; Higa, R.H.; Rosa, A.N.; Siqueira, F.; Silva, L.O.C.; Junior, R.A.A.T.; Coutinho, L.; Mudadu, M.A.; Alencar, M.; et al. A genome-wide scan for selection signatures in Nellore cattle. Anim. Genet. 2014, 45, 771–781. [Google Scholar] [CrossRef]
- Gautier, M.; Flori, L.; Riebler, A.; Jaffrézic, F.; Laloé, D.; Gut, I.; Moazami-Goudarzi, K.; Foulley, J.-L. A whole genome Bayesian scan for adaptive genetic divergence in West African cattle. BMC Genom. 2009, 10, 550. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hanotte, O.; Mwai, O.A.; Dessie, T.; Bashir, S.; Diallo, B.; Agaba, M.; Kim, K.; Kwak, W.; Sung, S.; et al. The genome landscape of indigenous African cattle. Genome Biol. 2017, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39 (Suppl. 2), W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [Green Version]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Sudhir, K.; Glen, S.; Koichiro, T. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.J.; Clark, A.G.; Bustamante, C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGiorgio, M.; Huber, C.D.; Hubisz, M.; Hellmann, I.; Nielsen, R. SweepFinder2: Increased sensitivity, robustness and flexibility. Bioinformatics 2016, 32, 1895–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Patterson, N.; Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 2010, 20, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara-Miki, R.; Tsuda, K.; Shiwa, Y.; Arai-Kichise, Y.; Matsumoto, T.; Kanesaki, Y.; Oda, S.-I.; Ebihara, S.; Yajima, S.; Yoshikawa, H.; et al. Whole-genome resequencing shows numerous genes with nonsynonymous SNPs in the Japanese native cattle Kuchinoshima-Ushi. BMC Genom. 2011, 12, 103. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Zhang, K.; Hong, Y.; Wang, J.; Liu, Q.; Zhang, Z.; Xia, H.; Tang, Y.; Li, T.; Li, L.; et al. miR-342-3p promotes osteogenic differentiation via targetingATF3. BMC Genom. 2018, 592, 4051–4065. [Google Scholar] [CrossRef] [Green Version]
- Bacchelli, E.; Loi, E.; Cameli, C.; Moi, L.; Benedetti, A.F.V.; Blois, S.; Fadda, A.; Bonora, E.; Mattu, S.; Fadda, R.; et al. Analysis of a Sardinian Multiplex Family with Autism Spectrum Disorder Points to Post-Synaptic Density Gene Variants and Identifies CAPG as a Functionally Relevant Candidate Gene. J. Clin. Med. 2019, 8, 212. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhang, J.; Liu, B.; Huang, Y.; Li, S.; Wen, H.; Zhang, M.; Li, J.; Li, Y.; He, F. Identification and Characterization of lncRNAs Related to the Muscle Growth and Development of Japanese Flounder (Paralichthys olivaceus). Front. Genet. 2020, 11, 1034. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, Y.; Gu, L.; Lan, M.; Liu, C.; Wang, M.; Su, Y.; Ge, M.; Wang, T.; Yu, Y.; et al. Islr regulates canonical Wnt signaling-mediated skeletal muscle regeneration by stabilizing Dishevelled-2 and preventing autophagy. Nat. Commun. 2018, 9, 5129. [Google Scholar] [CrossRef] [Green Version]
- Fontanesi, L.; Colombo, M.; Tognazzi, L.; Scotti, E.; Buttazzoni, L.; Dall’Olio, S.; Davoli, R.; Russo, V. The porcine TBC1D1 gene: Mapping, SNP identification, and association study with meat, carcass and production traits in Italian heavy pigs. Mol. Biol. Rep. 2010, 38, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhang, S.; Zhang, H.; Zhang, Z.; Chen, N.; Li, Z.; Sun, H.; Liu, X.; Lyu, S.; Wang, X.; et al. Assessing genomic diversity and signatures of selection in Jiaxian Red cattle using whole-genome sequencing data. BMC Genom. 2021, 22, 43. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Q.; Xu, S.; Jin, P.; Luan, P.; Li, Y.; Cao, Z.; Leng, L.; Wang, Y.; Wang, S. Differential expression of six chicken genes associated with fatness traits in a divergently selected broiler population. Mol. Cell. Probes 2016, 30, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Liu, X.; Gebreselassie, G.; Abied, A.; Ma, Q.; Ma, Y. Genome-wide association analysis reveals the genetic locus for high reproduction trait in Chinese Arbas Cashmere goat. Genes Genom. 2020, 42, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, M.; Zhou, M.; Wang, Y.; Wu, X.; Zhang, X.; Ding, Y.; Zhao, G.; Yin, Z.; Wang, C. Identification of Signatures of Selection by Whole-Genome Resequencing of a Chinese Native Pig. Front. Genet. 2020, 11, 566255. [Google Scholar] [CrossRef] [PubMed]
- Makina, S.O.; Muchadeyi, F.C.; Van Marle-Köster, E.; Taylor, J.F.; Makgahlela, M.L.; Maiwashe, A. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Sel. Evol. 2015, 47, 92. [Google Scholar] [CrossRef] [Green Version]
- Comte, D.; Karampetsou, M.P.; Humbel, M.; Tsokos, G.C. Signaling lymphocyte activation molecule family in systemic lupus erythematosus. Clin. Immunol. 2018, 204, 57–63. [Google Scholar] [CrossRef]
- Van Driel, B.J.; Eliao, G.; Eengel, P.; Eterhorst, C. Responses to Microbial Challenges by SLAMF Receptors. Front. Immunol. 2016, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- García-Borrón, J.C.; Abdel-Malek, Z.; Jiménez-Cervantes, C. MC1R, the cAMP pathway, and the response to solar UV: Extending the horizon beyond pigmentation. Pigment Cell Melanoma Res. 2014, 27, 699–720. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, H.; Lai, W.; Hu, M.; Zhang, Y.; Bai, C.; Liu, J.; Ren, H.; Li, F.; Yan, S. Genome-wide re-sequencing reveals population structure and genetic diversity of Bohai Black cattle. Anim. Genet. 2021, 53, 133–136. [Google Scholar] [CrossRef]
- Notter, D.R. The importance of genetic diversity in livestock populations of the future. J. Anim. Sci. 1999, 77, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sun, F.Z.; Zhu, F.X.; Feng, H.Y.; Han, X. Runs of homozygosity and its application on livestock genome study. Hereditas 2019, 41, 304–317. (In Chinese) [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Liu, X.; Yang, T.; Huang, Z.; Hanif, Q.; Asif, M.; Khan, Q.M.; Mansoor, S. Genomic variants identified from whole-genome resequencing of indicine cattle breeds from Pakistan. PLoS ONE 2019, 14, e0215065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qanbari, S.; Pausch, H.; Jansen, S.; Somel, M.; Strom, T.M.; Fries, R.; Nielsen, R.; Simianer, H. Classic Selective Sweeps Revealed by Massive Sequencing in Cattle. PLoS Genet. 2014, 10, e1004148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, A.; Stapleton, P.; Corcoran, D.; Hamill, R.; White, A. Understanding meat quality through the application of genomic and proteomic approaches. Meat Sci. 2006, 74, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Ichigotani, Y.; Fujii, K.; Hamaguchi, M.; Matsuda, S. In search of a function for the E3B1/Abi2/Argbp1/NESH family (Review). Int. J. Mol. Med. 2002, 9, 591–595. [Google Scholar] [CrossRef]
- Dokas, J.; Chadt, A.; Nolden, T.; Himmelbauer, H.; Zierath, J.; Joost, H.-G.; Al-Hasani, H. Conventional Knockout of Tbc1d1 in Mice Impairs Insulin- and AICAR-Stimulated Glucose Uptake in Skeletal Muscle. Endocrinol. 2013, 154, 3502–3514. [Google Scholar] [CrossRef]
- Deng, B.; Parthasarathy, S.; Wang, W.; Gibney, B.R.; Battaile, K.P.; Lovell, S.; Benson, D.R.; Zhu, H. Study of the Individual Cytochrome b5 and Cytochrome b5 Reductase Domains of Ncb5or Reveals a Unique Heme Pocket and a Possible Role of the CS Domain*. J. Biol. Chem. 2010, 285, 30181–30191. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, F.; Kigoshi, H.; Fukushima, M.; Iwamoto, E.; Kobayashi, E.; Oyama, K.; Mannen, H.; Sasazaki, S. Whole-genome resequencing to identify candidate genes for the QTL for oleic acid percentage in Japanese Black cattle. Anim. Sci. J. 2019, 90, 467–472. [Google Scholar] [CrossRef]
- Rees, J.L. The Genetics of Sun Sensitivity in Humans. Am. J. Hum. Genet. 2004, 75, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Cui, R.; Widlund, H.R.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central Role of p53 in the Suntan Response and Pathologic Hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Malek, Z.A.; Swope, V.B.; Starner, R.J.; Koikov, L.; Cassidy, P.; Leachman, S. Melanocortins and the melanocortin 1 receptor, moving translationally towards melanoma prevention. Arch. Biochem. Biophys. 2014, 563, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, T.; Chen, C.; Ichikawa, R.; Zheng, Q.; Pei, L.; Takemura, I.; Nsobi, L.H.; Tabata, H.; Pan, H.; et al. Whole-genome sequencing of endangered Zhoushan cattle suggests its origin and the association of MC1R with black coat colour. Sci. Rep. 2021, 11, 17359. [Google Scholar] [CrossRef]
- Liu, R.; Jin, L.; Long, K.; Chai, J.; Ma, J.; Tang, Q.; Tian, S.; Hu, Y.; Lin, L.; Wang, X.; et al. Detection of genetic diversity and selection at the coding region of the melanocortin receptor 1 (MC1R) gene in Tibetan pigs and Landrace pigs. Gene 2015, 575, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, F.; Cao, J.; Liu, X. Skin Transcriptome Profiles Associated with Skin Color in Chickens. PLoS ONE 2015, 10, e0127301. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Cheng, H.; Liu, Y.; Sun, L.; Chen, N.; Jiang, F.; You, W.; Yang, Z.; Zhang, B.; Song, E.; et al. Assessing Genomic Diversity and Selective Pressures in Bohai Black Cattle Using Whole-Genome Sequencing Data. Animals 2022, 12, 665. https://doi.org/10.3390/ani12050665
Ma X, Cheng H, Liu Y, Sun L, Chen N, Jiang F, You W, Yang Z, Zhang B, Song E, et al. Assessing Genomic Diversity and Selective Pressures in Bohai Black Cattle Using Whole-Genome Sequencing Data. Animals. 2022; 12(5):665. https://doi.org/10.3390/ani12050665
Chicago/Turabian StyleMa, Xiaohui, Haijian Cheng, Yangkai Liu, Luyang Sun, Ningbo Chen, Fugui Jiang, Wei You, Zhangang Yang, Baoheng Zhang, Enliang Song, and et al. 2022. "Assessing Genomic Diversity and Selective Pressures in Bohai Black Cattle Using Whole-Genome Sequencing Data" Animals 12, no. 5: 665. https://doi.org/10.3390/ani12050665
APA StyleMa, X., Cheng, H., Liu, Y., Sun, L., Chen, N., Jiang, F., You, W., Yang, Z., Zhang, B., Song, E., & Lei, C. (2022). Assessing Genomic Diversity and Selective Pressures in Bohai Black Cattle Using Whole-Genome Sequencing Data. Animals, 12(5), 665. https://doi.org/10.3390/ani12050665