
Unravelling the Diversity and Abundance of the Red Fox (Vulpes vulpes) Faecal Resistome and the Phenotypic Antibiotic Susceptibility of Indicator Bacteria

 , ,
, ,  ,
,  , and
, and
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Study Areas and Sampling
2.2. DNA Extraction and Pooling
2.3. High-Throughput qPCR
2.4. Isolation and Selection of E. coli and Enterococcus spp.
2.5. Antibiotic Susceptibility Testing (AST)
2.6. Data Analysis
3. Results and Discussion
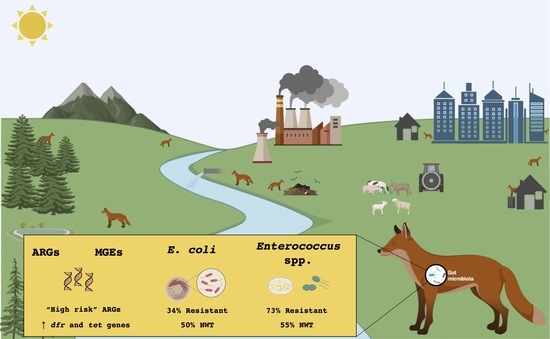
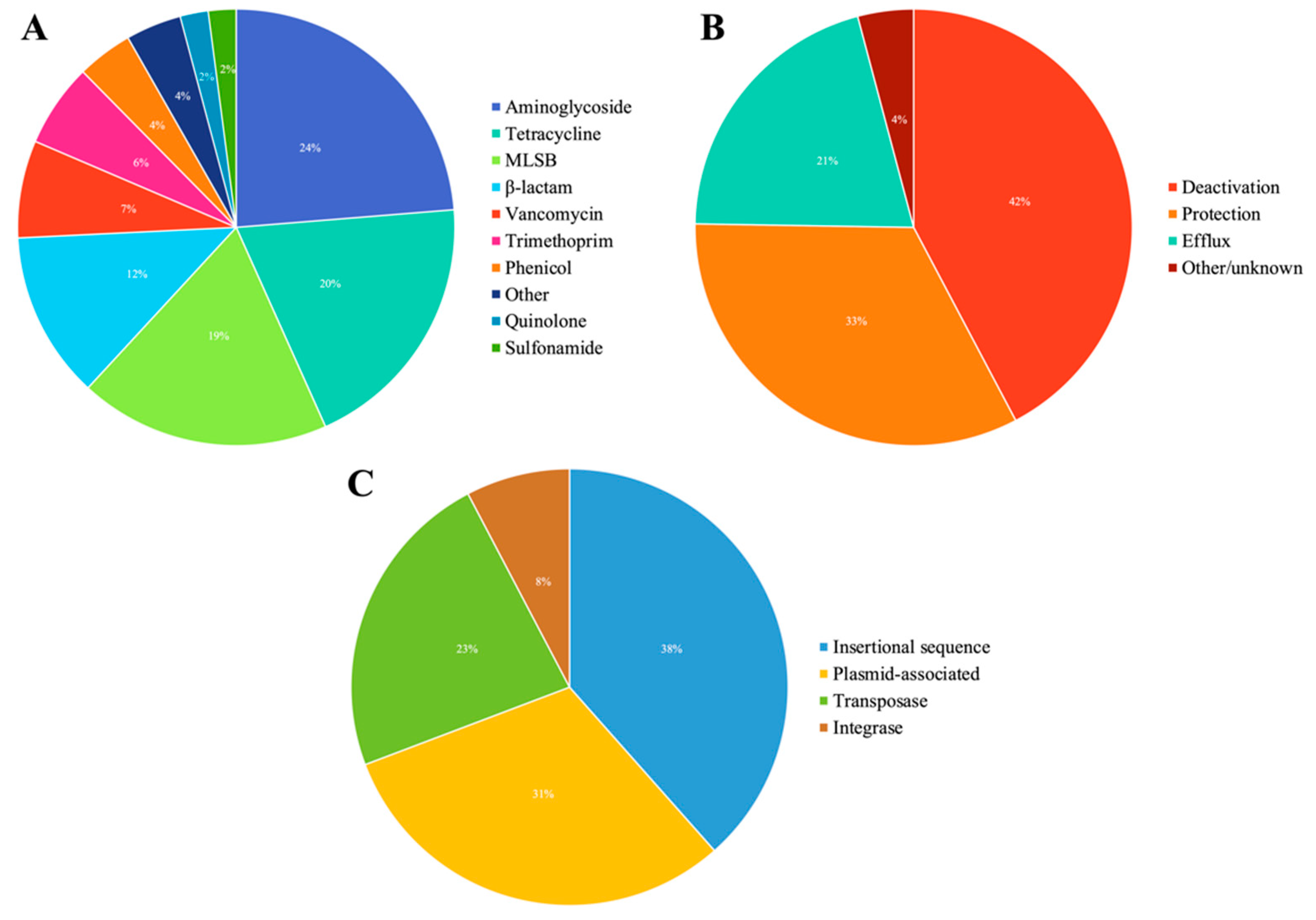
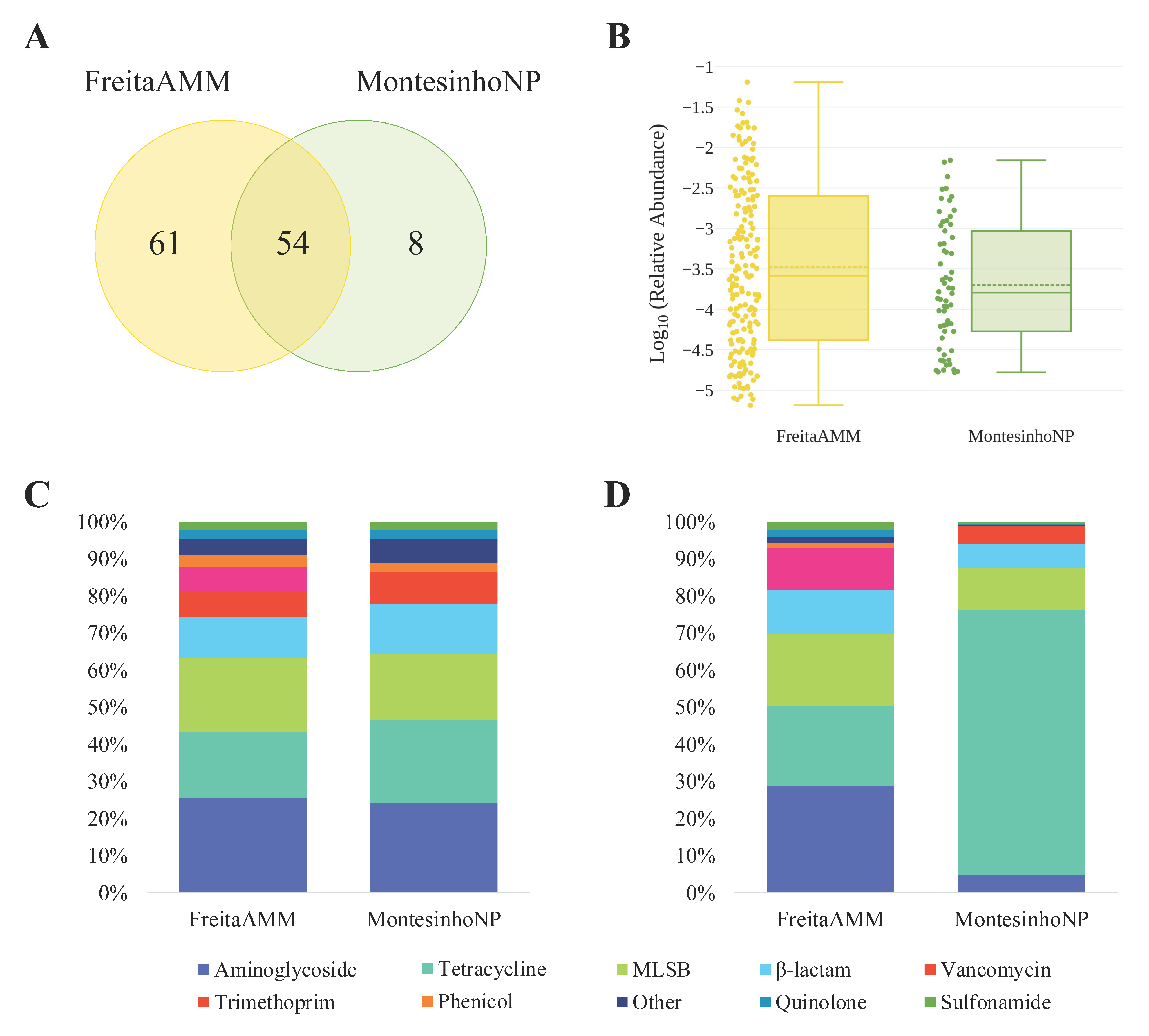
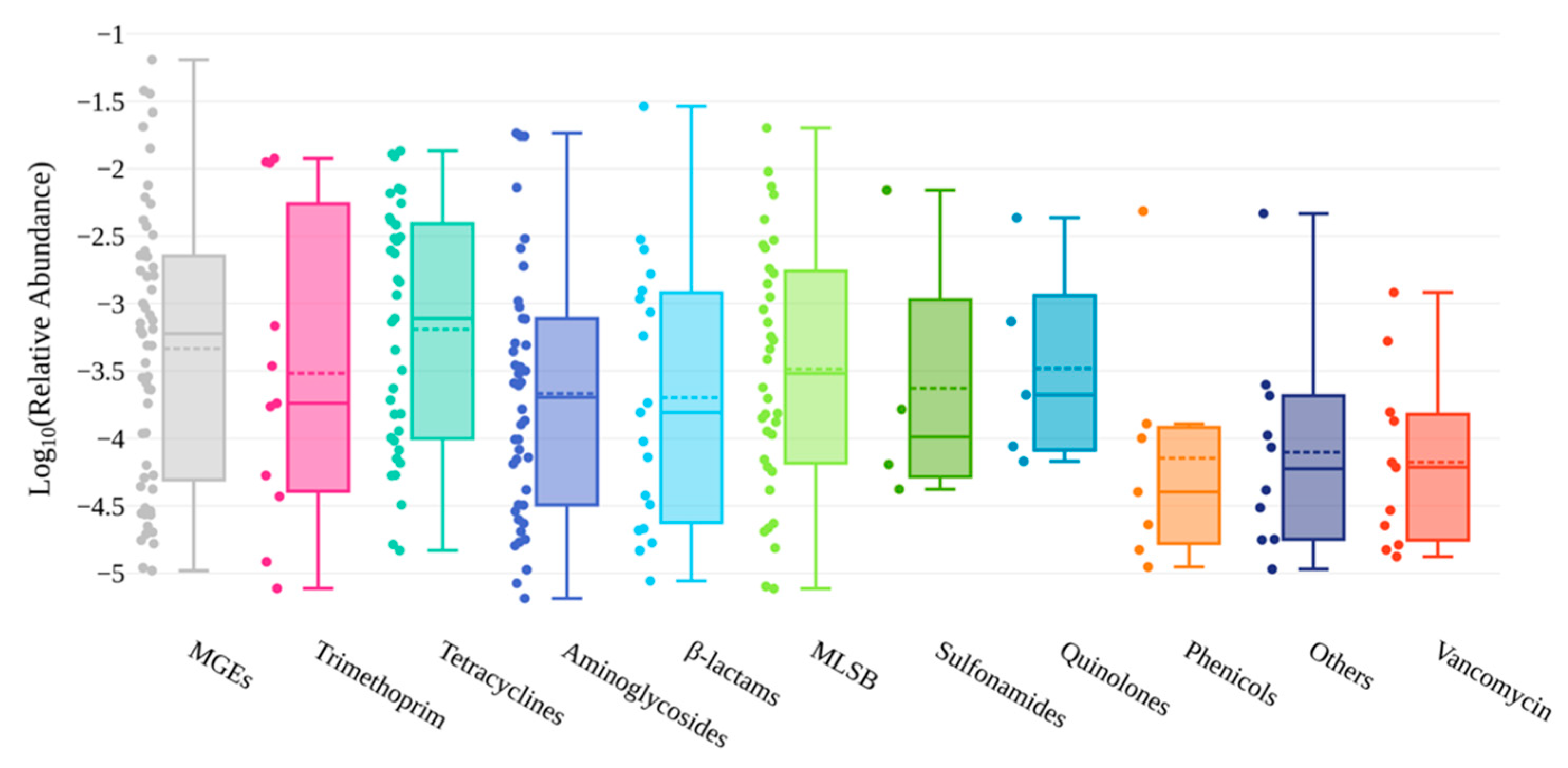
3.1. Overview of ARGs and MGEs Found in Red Fox
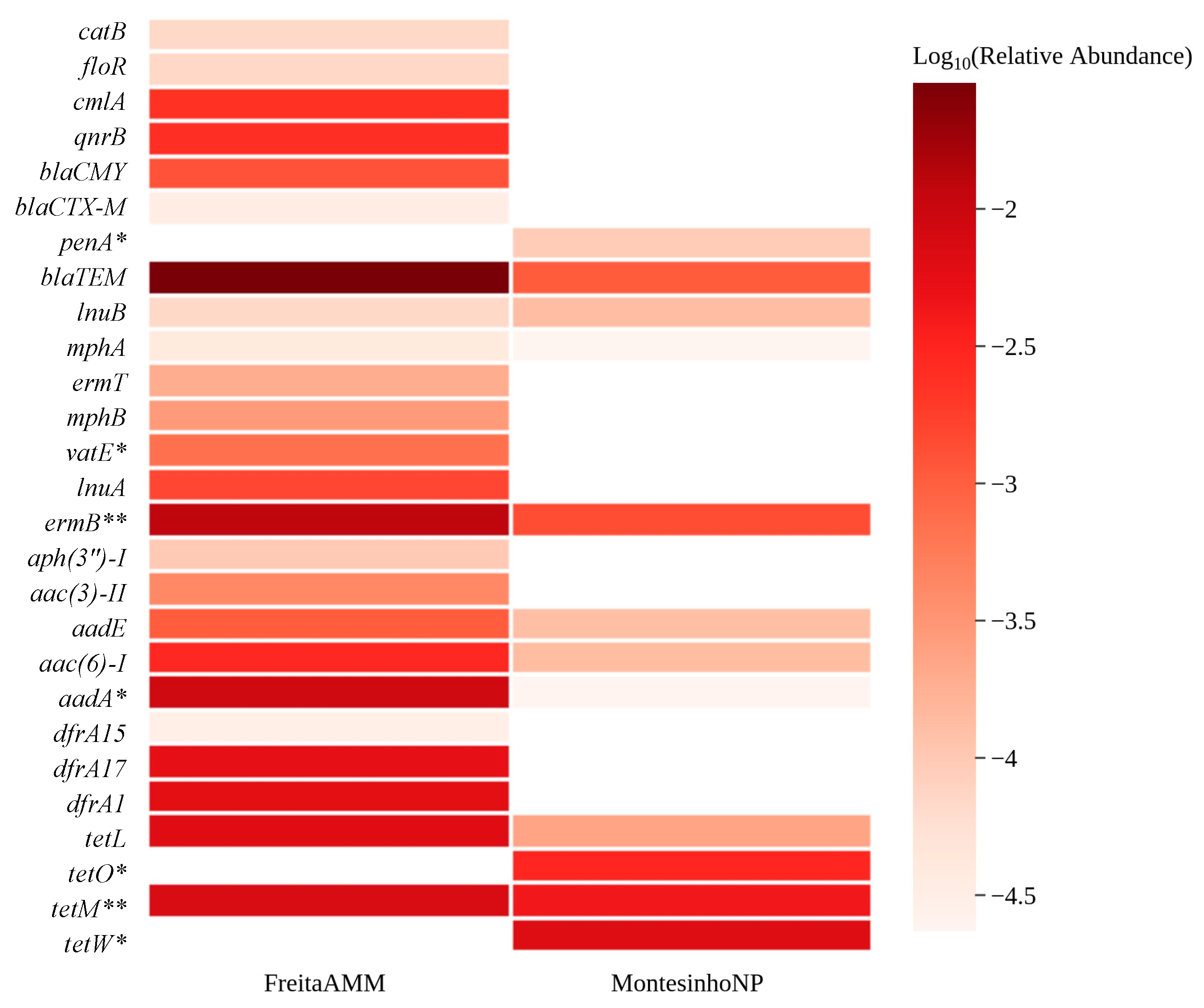
3.2. Abundance of AMR Genetic Determinants in Red Fox
3.3. Environmental Indicators and ARGs Associated with Human Health
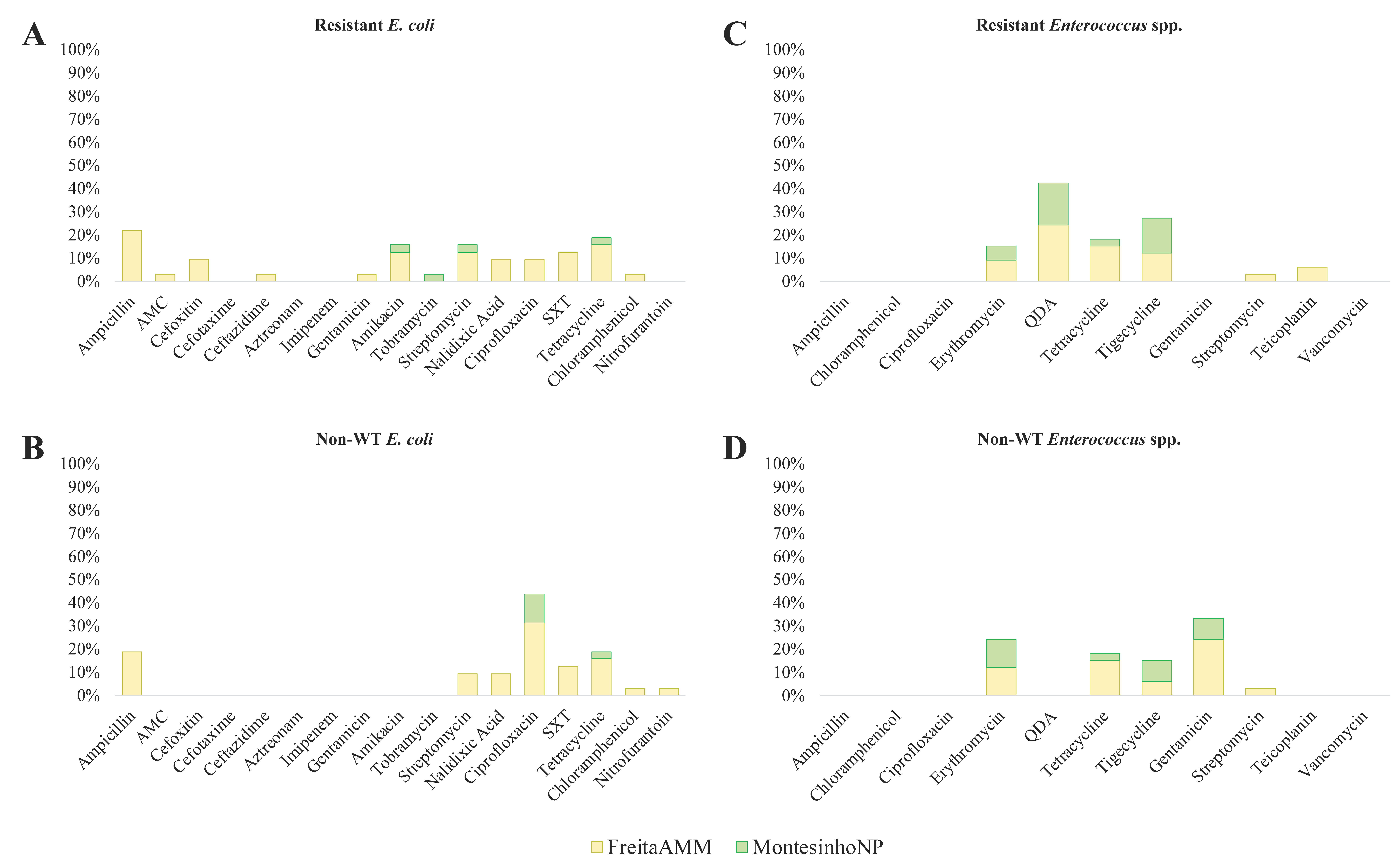
3.4. Prevalence of Antibiotic Resistance in E. coli
3.5. Prevalence of Antibiotic Resistance in Enterococcus spp.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jasovský, D.; Littmann, J.; Zorzet, A.; Cars, O. Antimicrobial Resistance—A Threat to the World’s Sustainable Development. Ups. J. Med. Sci. 2016, 121, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- WHO. Global Action Plan on Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Wright, G.D. Environmental and Clinical Antibiotic Resistomes, Same Only Different. Curr. Opin. Microbiol. 2019, 51, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.; Zembower, T.R. Antimicrobial Resistance. Gastrointest. Endosc. Clin. N. Am. 2020, 30, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.R.; Bonomo, R.A. Overview: The Ongoing Threat of Antimicrobial Resistance. Infect. Dis. Clin. N. Am. 2020, 34, 649–658. [Google Scholar] [CrossRef]
- Samreen; Ahmad, I.; Malak, H.A.; Abulreesh, H.H. Environmental Antimicrobial Resistance and Its Drivers: A Potential Threat to Public Health. J. Glob. Antimicrob. Resist. 2021, 27, 101–111. [Google Scholar] [CrossRef]
- Muurinen, J.; Stedtfeld, R.; Karkman, A.; Pärnänen, K.; Tiedje, J.; Virta, M. Influence of Manure Application on the Environmental Resistome under Finnish Agricultural Practice with Restricted Antibiotic Use. Environ. Sci. Technol. 2017, 51, 5989–5999. [Google Scholar] [CrossRef]
- Berendsen, B.J.A.; Wegh, R.S.; Memelink, J.; Zuidema, T.; Stolker, L.A.M. The Analysis of Animal Faeces as a Tool to Monitor Antibiotic Usage. Talanta 2015, 132, 258–268. [Google Scholar] [CrossRef]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the Wild: Antibiotic Resistance Genes in Natural Environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef]
- Vittecoq, M.; Godreuil, S.; Prugnolle, F.; Durand, P.; Brazier, L.; Renaud, N.; Arnal, A.; Aberkane, S.; Jean-Pierre, H.; Gauthier-Clerc, M.; et al. Antimicrobial Resistance in Wildlife. J. Appl. Ecol. 2016, 53, 519–529. [Google Scholar] [CrossRef]
- Torres, R.T.; Carvalho, J.; Cunha, M.V.; Serrano, E.; Palmeira, J.D.; Fonseca, C. Temporal and Geographical Research Trends of Antimicrobial Resistance in Wildlife—A Bibliometric Analysis. One Health 2020, 11, 100198. [Google Scholar] [CrossRef] [PubMed]
- Swift, B.M.C.; Bennett, M.; Waller, K.; Dodd, C.; Murray, A.; Gomes, R.L.; Humphreys, B.; Hobman, J.L.; Jones, M.A.; Whitlock, S.E.; et al. Anthropogenic Environmental Drivers of Antimicrobial Resistance in Wildlife. Sci. Total Environ. 2019, 649, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Sgroi, G.; Iatta, R.; Veneziano, V.; Bezerra-Santos, M.A.; Lesiczka, P.; Hrazdilová, K.; Annoscia, G.; D’Alessio, N.; Golovchenko, M.; Rudenko, N.; et al. Molecular Survey on Tick-Borne Pathogens and Leishmania Infantum in Red Foxes (Vulpes vulpes) from Southern Italy. Ticks Tick-Borne Dis. 2021, 12, 101669. [Google Scholar] [CrossRef] [PubMed]
- Dell’Arte, G.L.; Laaksonen, T.; Norrdahl, K.; Korpimäki, E. Variation in the Diet Composition of a Generalist Predator, the Red Fox, in Relation to Season and Density of Main Prey. Acta Oecol. 2007, 31, 276–281. [Google Scholar] [CrossRef]
- Karamon, J.; Dąbrowska, J.; Kochanowski, M.; Samorek-Pieróg, M.; Sroka, J.; Różycki, M.; Bilska-Zając, E.; Zdybel, J.; Cencek, T. Prevalence of Intestinal Helminths of Red Foxes (Vulpes vulpes) in Central Europe (Poland): A Significant Zoonotic Threat. Parasit. Vectors 2018, 11, 436. [Google Scholar] [CrossRef]
- Kaspersen, H.; Urdahl, A.M.; Simm, R.; Slettemeås, J.S.; Lagesen, K.; Norström, M. Occurrence of Quinolone Resistant E. Coli Originating from Different Animal Species in Norway. Vet. Microbiol. 2018, 217, 25–31. [Google Scholar] [CrossRef]
- O’Hagan, M.J.H.; Pascual-Linaza, A.V.; Couzens, C.; Holmes, C.; Bell, C.; Spence, N.; Huey, R.J.; Murphy, J.A.; Devaney, R.; Lahuerta-Marin, A. Estimation of the Prevalence of Antimicrobial Resistance in Badgers (Meles meles) and Foxes (Vulpes vulpes) in Northern Ireland. Front. Microbiol. 2021, 12, 596891. [Google Scholar] [CrossRef]
- Mo, S.S.; Urdahl, A.M.; Madslien, K.; Sunde, M.; Nesse, L.L.; Slettemeås, J.S.; Norström, M. What Does the Fox Say? Monitoring Antimicrobial Resistance in the Environment Using Wild Red Foxes as an Indicator. PLoS ONE 2018, 13, e0198019. [Google Scholar] [CrossRef]
- Turchi, B.; Dec, M.; Bertelloni, F.; Winiarczyk, S.; Gnat, S.; Bresciani, F.; Viviani, F.; Cerri, D.; Fratini, F. Antibiotic Susceptibility and Virulence Factors in Escherichia Coli from Sympatric Wildlife of the Apuan Alps Regional Park (Tuscany, Italy). Microb. Drug Resist. 2019, 25, 772–780. [Google Scholar] [CrossRef]
- Radhouani, H.; Igrejas, G.; Gonçalves, A.; Pacheco, R.; Monteiro, R.; Sargo, R.; Brito, F.; Torres, C.; Poeta, P. Antimicrobial Resistance and Virulence Genes in Escherichia Coli and Enterococci from Red Foxes (Vulpes vulpes). Anaerobe 2013, 23, 82–86. [Google Scholar] [CrossRef]
- Alonso, C.A.; Alcalá, L.; Simón, C.; Torres, C. Novel Sequence Types of Extended-Spectrum and Acquired AmpC Beta-Lactamase Producing Escherichia Coli and Escherichia Clade V Isolated from Wild Mammals. FEMS Microbiol. Ecol. 2017, 93, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Radhouani, H.; Igrejas, G.; Carvalho, C.; Pinto, L.; Gonçalves, A.; Lopez, M.; Sargo, R.; Cardoso, L.; Martinho, A.; Rego, V.; et al. Clonal Lineages, Antibiotic Resistance and Virulence Factors in Vancomycin-Resistant Enterococci Isolated from Fecal Samples of Red Foxes (Vulpes vulpes). J. Wildl. Dis. 2011, 47, 769–773. [Google Scholar] [CrossRef] [PubMed]
- McLain, J.E.; Cytryn, E.; Durso, L.M.; Young, S. Culture-Based Methods for Detection of Antibiotic Resistance in Agroecosystems: Advantages, Challenges, and Gaps in Knowledge. J. Environ. Qual. 2016, 45, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Stedtfeld, R.D.; Guo, X.; Stedtfeld, T.M.; Sheng, H.; Williams, M.R.; Hauschild, K.; Gunturu, S.; Tift, L.; Wang, F.; Howe, A.; et al. Primer Set 2.0 for Highly Parallel QPCR Array Targeting Antibiotic Resistance Genes and Mobile Genetic Elements. FEMS Microbiol. Ecol. 2018, 94, fiy130. [Google Scholar] [CrossRef]
- WHO. Integrated Surveillance of Antimicrobial Resistance in Foodborne Bacteria: Application of a One Health Approach; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Berendonk, T.U.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; Cytryn, E.; Walsh, F.; Bürgmann, H.; Sørum, H.; Norström, M.; Pons, M.-N.; et al. Tackling Antibiotic Resistance: The Environmental Framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef]
- Dias, D.; Fonseca, C.; Mendo, S.; Caetano, T. A Closer Look on the Variety and Abundance of the Faecal Resistome of Wild Boar. Environ. Pollut. 2022, 292, 118406. [Google Scholar] [CrossRef]
- Furstenau, T.N.; Cocking, J.H.; Hepp, C.M.; Fofanov, V.Y. Sample Pooling Methods for Efficient Pathogen Screening: Practical Implications. PLoS ONE 2020, 15, e0236849. [Google Scholar] [CrossRef]
- Zaheer, R.; Cook, S.R.; Barbieri, R.; Goji, N.; Cameron, A.; Petkau, A.; Polo, R.O.; Tymensen, L.; Stamm, C.; Song, J.; et al. Surveillance of Enterococcus Spp. Reveals Distinct Species and Antimicrobial Resistance Diversity across a One-Health Continuum. Sci. Rep. 2020, 10, 3937. [Google Scholar] [CrossRef]
- Singh, K.V.; Weinstock, G.M.; Murray, B.E. An Enterococcus Faecalis ABC Homologue (Lsa) Is Required for the Resistance of This Species to Clindamycin and Quinupristin-Dalfopristin. Antimicrob. Agents Chemother. 2002, 46, 1845–1850. [Google Scholar] [CrossRef]
- Kronvall, G.; Smith, P. Normalized Resistance Interpretation, the NRI Method. APMIS 2016, 124, 1023–1030. [Google Scholar] [CrossRef]
- Sievert, C. Interactive Web-Based Data Visualization with R, Plotly, and Shiny; Chapman and Hall/CRC: New York, NY, USA, 2020; ISBN 9780429447273. [Google Scholar]
- Radhouani, H.; Igrejas, G.; Gonçalves, A.; Estepa, V.; Sargo, R.; Torres, C.; Poeta, P. Molecular Characterization of Extended-Spectrum-Beta-Lactamase-Producing Escherichia Coli Isolates from Red Foxes in Portugal. Arch. Microbiol. 2013, 195, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Skarżyńska, M.; Leekitcharoenphon, P.; Hendriksen, R.S.; Aarestrup, F.M.; Wasyl, D. A Metagenomic Glimpse into the Gut of Wild and Domestic Animals: Quantification of Antimicrobial Resistance and More. PLoS ONE 2020, 15, e0242987. [Google Scholar] [CrossRef]
- Muurinen, J.; Richert, J.; Wickware, C.L.; Richert, B.; Johnson, T.A. Swine Growth Promotion with Antibiotics or Alternatives Can Increase Antibiotic Resistance Gene Mobility Potential. Sci. Rep. 2021, 11, 5485. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.; Fonseca, C.; Caetano, T.; Mendo, S. Oh, Deer! How Worried Should We Be about the Diversity and Abundance of the Faecal Resistome of Red Deer? Sci. Total Environ. 2022, 825, 153831. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, W.; Yang, L.; Stedtfeld, R.D.; Peng, A.; Gu, C.; Boyd, S.A.; Li, H. Antibiotic Resistance Genes and Bacterial Communities in Cornfield and Pasture Soils Receiving Swine and Dairy Manures. Environ. Pollut. 2019, 248, 947–957. [Google Scholar] [CrossRef] [PubMed]
- McCann, C.M.; Christgen, B.; Roberts, J.A.; Su, J.-Q.; Arnold, K.E.; Gray, N.D.; Zhu, Y.-G.; Graham, D.W. Understanding Drivers of Antibiotic Resistance Genes in High Arctic Soil Ecosystems. Environ. Int. 2019, 125, 497–504. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yuan, Q.; Mathieu, J.; Stadler, L.; Senehi, N.; Sun, R.; Alvarez, P.J.J. Antibiotic Resistance Genes from Livestock Waste: Occurrence, Dissemination, and Treatment. npj Clean Water 2020, 3, 4. [Google Scholar] [CrossRef]
- Cevidanes, A.; Esperón, F.; Di Cataldo, S.; Neves, E.; Sallaberry-Pincheira, N.; Millán, J. Antimicrobial Resistance Genes in Andean Foxes Inhabiting Anthropized Landscapes in Central Chile. Sci. Total Environ. 2020, 724, 138247. [Google Scholar] [CrossRef]
- Gillings, M.R.; Gaze, W.H.; Pruden, A.; Smalla, K.; Tiedje, J.M.; Zhu, Y.-G. Using the Class 1 Integron-Integrase Gene as a Proxy for Anthropogenic Pollution. ISME J. 2015, 9, 1269–1279. [Google Scholar] [CrossRef]
- Zhang, A.-N.; Gaston, J.M.; Dai, C.L.; Zhao, S.; Poyet, M.; Groussin, M.; Yin, X.; Li, L.-G.; van Loosdrecht, M.C.M.; Topp, E.; et al. An Omics-Based Framework for Assessing the Health Risk of Antimicrobial Resistance Genes. Nat. Commun. 2021, 12, 4765. [Google Scholar] [CrossRef]
- Silva, N.; Phythian, C.J.; Currie, C.; Tassi, R.; Ballingall, K.T.; Magro, G.; McNeilly, T.N.; Zadoks, R.N. Antimicrobial Resistance in Ovine Bacteria: A Sheep in Wolf’s Clothing? PLoS ONE 2020, 15, e0238708. [Google Scholar] [CrossRef] [PubMed]
- Oliveira de Araujo, G.; Huff, R.; Favarini, M.O.; Mann, M.B.; Peters, F.B.; Frazzon, J.; Guedes Frazzon, A.P. Multidrug Resistance in Enterococci Isolated From Wild Pampas Foxes (Lycalopex Gymnocercus) and Geoffroy’s Cats (Leopardus Geoffroyi) in the Brazilian Pampa Biome. Front. Vet. Sci. 2020, 7, 606377. [Google Scholar] [CrossRef] [PubMed]
- ECDC Trend of Antimicrobial Consumption by Country. Available online: https://ecdc.europa.eu/en/antimicrobial-consumption/database/trend-country (accessed on 2 May 2022).
- European Medicines Agency. Sales of Veterinary Antimicrobial Agents in 31 European Countries in 2019 and 2020; European Surveillance of Veterinary Antimicrobial Consumption; Publications Office of the European Union: Luxembourg, 2021; 130p. [Google Scholar]
- Cardoso, A.R.; Carneiro, L.P.T.; Cabral-Miranda, G.; Bachmann, M.F.; Sales, M.G.F. Employing Bacteria Machinery for Antibiotic Detection: Using DNA Gyrase for Ciprofloxacin Detection. Chem. Eng. J. 2021, 409, 128135. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias, D.; Hipólito, D.; Figueiredo, A.; Fonseca, C.; Caetano, T.; Mendo, S. Unravelling the Diversity and Abundance of the Red Fox (Vulpes vulpes) Faecal Resistome and the Phenotypic Antibiotic Susceptibility of Indicator Bacteria. Animals 2022, 12, 2572. https://doi.org/10.3390/ani12192572
Dias D, Hipólito D, Figueiredo A, Fonseca C, Caetano T, Mendo S. Unravelling the Diversity and Abundance of the Red Fox (Vulpes vulpes) Faecal Resistome and the Phenotypic Antibiotic Susceptibility of Indicator Bacteria. Animals. 2022; 12(19):2572. https://doi.org/10.3390/ani12192572
Chicago/Turabian StyleDias, Diana, Dário Hipólito, Ana Figueiredo, Carlos Fonseca, Tânia Caetano, and Sónia Mendo. 2022. "Unravelling the Diversity and Abundance of the Red Fox (Vulpes vulpes) Faecal Resistome and the Phenotypic Antibiotic Susceptibility of Indicator Bacteria" Animals 12, no. 19: 2572. https://doi.org/10.3390/ani12192572
APA StyleDias, D., Hipólito, D., Figueiredo, A., Fonseca, C., Caetano, T., & Mendo, S. (2022). Unravelling the Diversity and Abundance of the Red Fox (Vulpes vulpes) Faecal Resistome and the Phenotypic Antibiotic Susceptibility of Indicator Bacteria. Animals, 12(19), 2572. https://doi.org/10.3390/ani12192572