
One-Step In Vitro Generation of ETV2-Null Pig Embryos

 , , , , , ,
, , , , , ,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Pig Fibroblast Isolation
2.2. Guide RNA Design and In Vitro Testing
2.3. Surveyor Analysis
2.4. Oocyte Collection and In Vitro Maturation (IVM)
2.5. In Vitro Fertilization (IVF)
2.6. In Vivo Fertilization and Zygote Collection
2.7. Alt-R™ CRISPR/Cas9 Ribonucleoprotein Complex Formation
2.8. Microinjection of CRISPR/Cas9 in Porcine Zygotes
2.9. Embryo Culture
2.10. Genomic DNA Extraction, Library Preparation for Next-Generation Sequencing (NGS), and Indel Detection
2.11. Statistical Analysis
3. Results
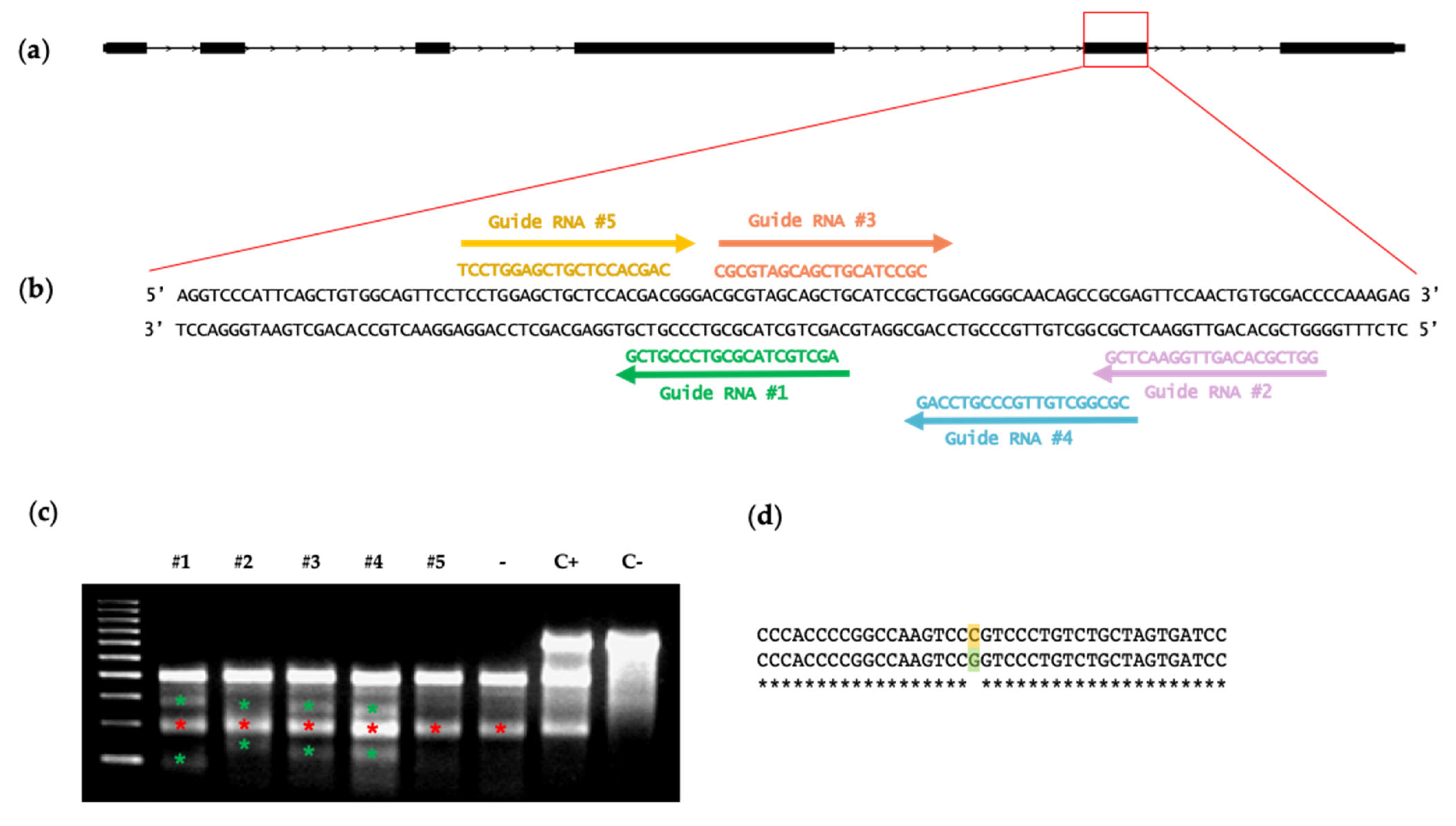
3.1. In Vitro Selection of Functional ETV2 Guide RNAs
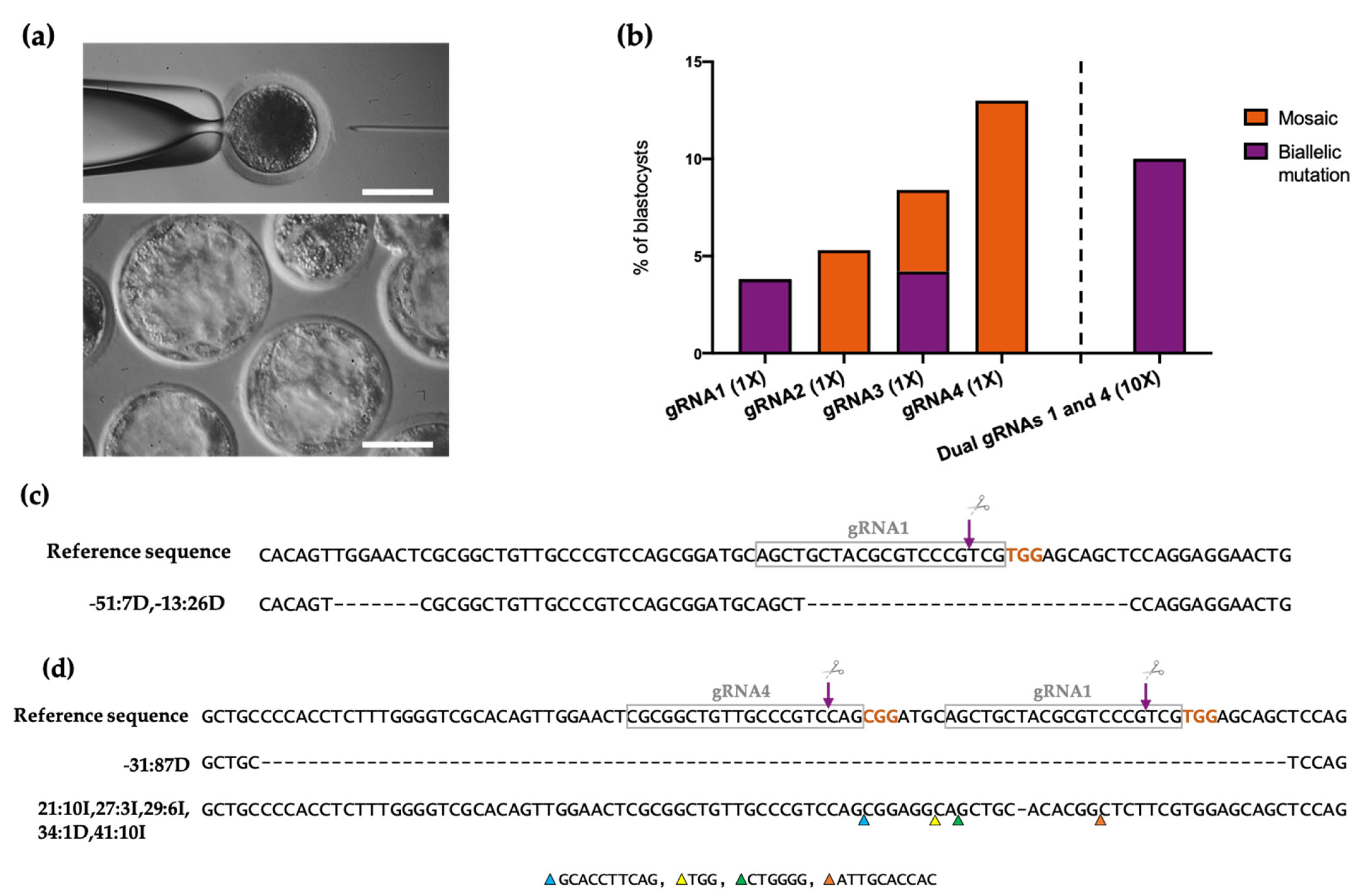
3.2. Production of ETV2-Mutated Pig Blastocysts with Single gRNAs
3.3. Improvement of the Mutation Rate with Dual gRNAs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kobayashi, T.; Yamaguchi, T.; Hamanaka, S.; Kato-Itoh, M.; Yamazaki, Y.; Ibata, M.; Sato, H.; Lee, Y.-S.; Usui, J.-I.; Knisely, A.; et al. Generation of Rat Pancreas in Mouse by Interspecific Blastocyst Injection of Pluripotent Stem Cells. Cell 2010, 142, 787–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, D.K. Potential Benefits and Risks of Clinical Xenotransplantation. Transpl. Res. Risk Manag. 2012, 4, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Matsunari, H.; Nagashima, H.; Watanabe, M.; Umeyama, K.; Nakano, K.; Nagaya, M.; Kobayashi, T.; Yamaguchi, T.; Sumazaki, R.; Herzenberg, L.A.; et al. Blastocyst Complementation Generates Exogenic Pancreas In Vivo in Apancreatic Cloned Pigs. Proc. Natl. Acad. Sci. USA 2013, 110, 4557–4562. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Vilarino, M.; Suzuki, K.; Okamura, D.; Bogliotti, Y.S.; Park, I.; Rowe, J.; McNabb, B.; Ross, P.J.; Belmonte, J.C.I. CRISPR-Cas9 Mediated One-Step Disabling of Pancreatogenesis in Pigs. Sci. Rep. 2017, 7, 10487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanihara, F.; Hirata, M.; Thi Nguyen, N.; Anh Le, Q.; Hirano, T.; Otoi, T. Generation of Viable PDX1 Gene-Edited Founder Pigs as Providers of Nonmosaics. Mol. Reprod. Dev. 2020, 87, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Matsunari, H.; Watanabe, M.; Hasegawa, K.; Uchikura, A.; Nakano, K.; Umeyama, K.; Masaki, H.; Hamanaka, S.; Yamaguchi, T.; Nagaya, M.; et al. Compensation of Disabled Organogeneses in Genetically Modified Pig Fetuses by Blastocyst Complementation. Stem Cell Rep. 2020, 14, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Nakano, K.; Uchikura, A.; Matsunari, H.; Yashima, S.; Umeyama, K.; Takayanagi, S.; Sakuma, T.; Yamamoto, T.; Morita, S.; et al. Anephrogenic Phenotype Induced by SALL1 Gene Knockout in Pigs. Sci. Rep. 2019, 9, 8016. [Google Scholar] [CrossRef]
- Cooper, D.K.; Ezzelarab, M.B.; Hara, H.; Iwase, H.; Lee, W.; Wijkstrom, M.; Bottino, R. The Pathobiology of Pig-to-Primate Xenotransplantation: A Historical Review. Xenotransplantation 2016, 23, 83–105. [Google Scholar] [CrossRef]
- Das, S.; Koyano-Nakagawa, N.; Gafni, O.; Maeng, G.; Singh, B.; Rasmussen, T.; Pan, X.; Choi, K.-D.; Mickelson, D.; Gong, W.; et al. Generation of Human Endothelium in Pig Embryos Deficient in ETV2. Nat. Biotechnol. 2020, 38, 297–302. [Google Scholar] [CrossRef]
- Liu, T.; Dou, H.; Xiang, X.; Lin, L.; Li, Y.; Pang, X.; Zhang, Y.; Chen, Y.; Luan, J.; Xu, Y.; et al. Factors Determining the Efficiency of Porcine Somatic Cell Nuclear Transfer: Data Analysis with Over 200,000 Reconstructed Embryos. Cell. Reprogramming 2015, 17, 463–471. [Google Scholar] [CrossRef]
- Carter, D.B.; Lai, L.; Park, K.-W.; Samuel, M.; Lattimer, J.C.; Jordan, K.R.; Estes, D.M.; Besch-Williford, C.; Prather, R.S. Phenotyping of Transgenic Cloned Piglets. Cloning Stem Cells 2002, 4, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-R.; Cho, S.-K.; Lee, S.-Y.; Choi, Y.-J.; Park, J.-Y.; Kwon, D.-N.; Son, W.-J.; Paik, S.-S.; Kim, T.; Han, Y.-M.; et al. A Rare and Often Unrecognized Cerebromeningitis and Hemodynamic Disorder: A Major Cause of Sudden Death in Somatic Cell Cloned Piglets. Proteomics 2005, 5, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, K.M.; Lee, K.; Benne, J.A.; Beaton, B.P.; Spate, L.D.; Murphy, S.L.; Samuel, M.S.; Mao, J.; O’Gorman, C.; Walters, E.M.; et al. Use of the CRISPR/Cas9 System to Produce Genetically Engineered Pigs from In Vitro-Derived Oocytes and Embryos1. Biol. Reprod. 2014, 91, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Farrell, K.; Uh, K. Application of Genome-Editing Systems to Enhance Available Pig Resources for Agriculture and Biomedicine. Reprod. Fertil. Dev. 2020, 32, 40–49. [Google Scholar] [CrossRef]
- Ryu, J.; Prather, R.S.; Lee, K. Use of gene-editing technology to introduce targeted modifications in pigs. J. Anim. Sci. Biotechnol. 2018, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Hai, T.; Teng, F.; Guo, R.; Li, W.; Zhou, Q. One-Step Generation of Knockout Pigs by Zygote Injection of CRISPR/Cas System. Cell Res. 2014, 24, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Wareing, S.; Mazan, A.; Pearson, S.; Göttgens, B.; Lacaud, G.; Kouskoff, V. The Flk1-Cre-Mediated Deletion of ETV2 Defines Its Narrow Temporal Requirement During Embryonic Hematopoietic Development. Stem Cells 2012, 30, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Ferdous, A.; Caprioli, A.; Iacovino, M.; Martin, C.M.; Morris, J.; Richardson, J.A.; Latif, S.; Hammer, R.E.; Harvey, R.P.; Olson, E.N.; et al. Nkx2–5 Transactivates the Ets-Related Protein 71 Gene and Specifies an Endothelial/Endocardial Fate in the Developing Embryo. Proc. Natl. Acad. Sci. USA 2009, 106, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Petters, R.M.; Wells, K.D. Culture of Pig Embryos. J. Reprod. Fertil. Suppl. 1993, 48, 61–73. [Google Scholar] [CrossRef]
- Funahashi, H.; Cantley, T.C.; Day, B.N. Synchronization of Meiosis in Porcine Oocytes by Exposure to Dibutyryl Cyclic Adenosine Monophosphate Improves Developmental Competence Following in Vitro Fertilization1. Biol. Reprod. 1997, 57, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pursel, V.G.; Johnson, L.A. Freezing of Boar Spermatozoa: Fertilizing Capacity with Concentrated Semen and a New Thawing Procedure. J. Anim. Sci. 1975, 40, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Martinez, E.A.; Angel, M.A.; Cuello, C.; Sanchez-Osorio, J.; Gomis, J.; Parrilla, I.; Vila, J.; Colina, I.; Diaz, M.; Reixach, J.; et al. Successful Non-Surgical Deep Uterine Transfer of Porcine Morulae after 24 Hour Culture in a Chemically Defined Medium. PLoS ONE 2014, 9, e104696. [Google Scholar] [CrossRef] [PubMed]
- Canovas, S.; Ivanova, E.; Romar, R.; García-Martínez, S.; Soriano-Úbeda, C.; García-Vázquez, A.F.; Saadeh, H.; Andrews, S.; Kelsey, G.; Coy, P. DNA Methylation and Gene Expression Changes Derived from Assisted Reproductive Technologies Can Be Decreased by Reproductive Fluids. eLife 2017, 6, e23670. [Google Scholar] [CrossRef]
- Lindsay, H.; Burger, A.; Biyong, B.; Felker, A.; Hess, C.; Zaugg, J.; Chiavacci, E.; Anders, C.; Jinek, M.; Mosimann, C.; et al. CrispRVariants Charts the Mutation Spectrum of Genome Engineering Experiments. Nat. Biotechnol. 2016, 34, 701–702. [Google Scholar] [CrossRef]
- Brown, T.A.; McKnight, S.L. Specificities of Protein-Protein and Protein-DNA Interaction of GABP Alpha and Two Newly Defined Ets-Related Proteins. Genes Dev. 1992, 6, 2502–2512. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.W.; Kim, A. Deletion of transcription factor binding motifs using the CRISPR/spCas9 system in the β-globin LCR. Biosci. Rep. 2017, 37, BSR20170976. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Furuhashi, K.; Danielsson, J.A.; Hirata, Y.; Kakiuchi, M.; Lin, C.-S.; Ohta, M.; Riccio, P.; Takahashi, Y.; Xu, X.; et al. Generation of Functional Lungs via Conditional Blastocyst Complementation Using Pluripotent Stem Cells. Nat. Med. 2019, 25, 1691–1698. [Google Scholar] [CrossRef]
- Hamanaka, S.; Umino, A.; Sato, H.; Hayama, T.; Yanagida, A.; Mizuno, N.; Kobayashi, T.; Kasai, M.; Suchy, F.P.; Yamazaki, S.; et al. Generation of Vascular Endothelial Cells and Hematopoietic Cells by Blastocyst Complementation. Stem Cell Rep. 2018, 11, 988–997. [Google Scholar] [CrossRef] [Green Version]
- Usui, J.-I.; Kobayashi, T.; Yamaguchi, T.; Knisely, A.; Nishinakamura, R.; Nakauchi, H. Generation of Kidney from Pluripotent Stem Cells via Blastocyst Complementation. Am. J. Pathol. 2012, 180, 2417–2426. [Google Scholar] [CrossRef]
- Sandrin, M.S.; Vaughan, H.A.; Dabkowski, P.L.; McKenzie, I.F. Anti-pig IgM Antibodies in Human Serum React Predominantly with Gal (alpha 1-3) Gal epitopes. Proc. Natl. Acad. Sci. USA 1993, 90, 11391–11395. [Google Scholar] [CrossRef] [Green Version]
- Tisato, V.; Cozzi, E. Xenotransplantation: An Overview of the Field. Methods Mol. Biol. 2012, 885, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, K.E.; Mandawala, A.A.; Griffin, D.K.; Walling, G.A.; Harvey, S.C. The production of Pig Preimplantation Embryos In Vitro: Current Progress and Future Prospects. Reprod. Biol. 2018, 18, 203–211. [Google Scholar] [CrossRef]
- Tian, J.-H.; Wu, Z.-H.; Liu, L.; Cai, Y.; Zeng, S.-M.; Zhu, S.-E.; Liu, G.-S.; Li, Y.; Wu, C.-X. Effects of Oocyte Activation and Sperm Preparation on the Development of Porcine Embryos Derived from In Vitro-Matured Oocytes and Intracytoplasmic Sperm Injection. Theriogenology 2006, 66, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Tanihara, F.; Hirata, M.; Nguyen, N.T.; Le, Q.A.; Hirano, T.; Takemoto, T.; Nakai, M.; Fuchimoto, D.; Otoi, T. Generation of PDX -1 Mutant Porcine Blastocysts by Introducing CRISPR /Cas9-System into Porcine Zygotes via Electroporation. Anim. Sci. J. 2019, 90, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Yamashita, Y.; Takemoto, T. Electroporation of Cas9 Protein/sgRNA into Early Pronuclear Zygotes Generates Non-Mosaic Mutants in the Mouse. Dev. Biol. 2016, 418, 1–9. [Google Scholar] [CrossRef]
- Hirata, M.; Wittayarat, M.; Tanihara, F.; Sato, Y.; Namula, Z.; Le, Q.A.; Lin, Q.; Takebayashi, K.; Otoi, T. One-Step Genome Editing of Porcine Zygotes Through the Electroporation of a CRISPR/Cas9 System with Two Guide RNAs. In Vitro Cell Dev. Biol. 2020, 56, 614–621. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Putative Zygotes (n) | Cleavage Rates (%) | Blastocyst Rates (%) | Blastocysts Analyzed via NGS | |
|---|---|---|---|---|
| Control | 625 | 306/625 (48.96) | 70/306 (22.88) | 28 |
| gRNA1 | 144 | 86/144 (59.72) * | 30/86 (34.88) * | 26 |
| gRNA2 | 72 | 43/72 (59.72) | 26/43 (60.47) * | 19 |
| gRNA3 | 288 | 145/288 (50.35) | 31/145 (21.38) | 24 |
| gRNA4 | 291 | 141/291 (48.45) | 27/141 (19.15) | 23 |
| Putative Zygotes (n) | Cleavage Rates (%) | Blastocyst Rates (%) | Blastocysts Analyzed via NGS | |
|---|---|---|---|---|
| Control | 48 | 47/48 (97.92) | 21/47 (44.68) | 13 |
| gRNA1-4 5× | 20 | 17/20 (85.00) * | 9/17 (52.94) | 9 |
| gRNA1-4 10× | 62 | 33/62 (53.23) * | 10/33 (30.30) | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moya-Jódar, M.; Coppiello, G.; Rodríguez-Madoz, J.R.; Abizanda, G.; Barlabé, P.; Vilas-Zornoza, A.; Ullate-Agote, A.; Luongo, C.; Rodríguez-Tobón, E.; Navarro-Serna, S.; et al. One-Step In Vitro Generation of ETV2-Null Pig Embryos. Animals 2022, 12, 1829. https://doi.org/10.3390/ani12141829
Moya-Jódar M, Coppiello G, Rodríguez-Madoz JR, Abizanda G, Barlabé P, Vilas-Zornoza A, Ullate-Agote A, Luongo C, Rodríguez-Tobón E, Navarro-Serna S, et al. One-Step In Vitro Generation of ETV2-Null Pig Embryos. Animals. 2022; 12(14):1829. https://doi.org/10.3390/ani12141829
Chicago/Turabian StyleMoya-Jódar, Marta, Giulia Coppiello, Juan Roberto Rodríguez-Madoz, Gloria Abizanda, Paula Barlabé, Amaia Vilas-Zornoza, Asier Ullate-Agote, Chiara Luongo, Ernesto Rodríguez-Tobón, Sergio Navarro-Serna, and et al. 2022. "One-Step In Vitro Generation of ETV2-Null Pig Embryos" Animals 12, no. 14: 1829. https://doi.org/10.3390/ani12141829
APA StyleMoya-Jódar, M., Coppiello, G., Rodríguez-Madoz, J. R., Abizanda, G., Barlabé, P., Vilas-Zornoza, A., Ullate-Agote, A., Luongo, C., Rodríguez-Tobón, E., Navarro-Serna, S., París-Oller, E., Oficialdegui, M., Carvajal-Vergara, X., Ordovás, L., Prósper, F., García-Vázquez, F. A., & Aranguren, X. L. (2022). One-Step In Vitro Generation of ETV2-Null Pig Embryos. Animals, 12(14), 1829. https://doi.org/10.3390/ani12141829