
Breeding Sustainable Beef Cows: Reducing Weight and Increasing Productivity


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Genotypes
2.3. Genomic Heterosis
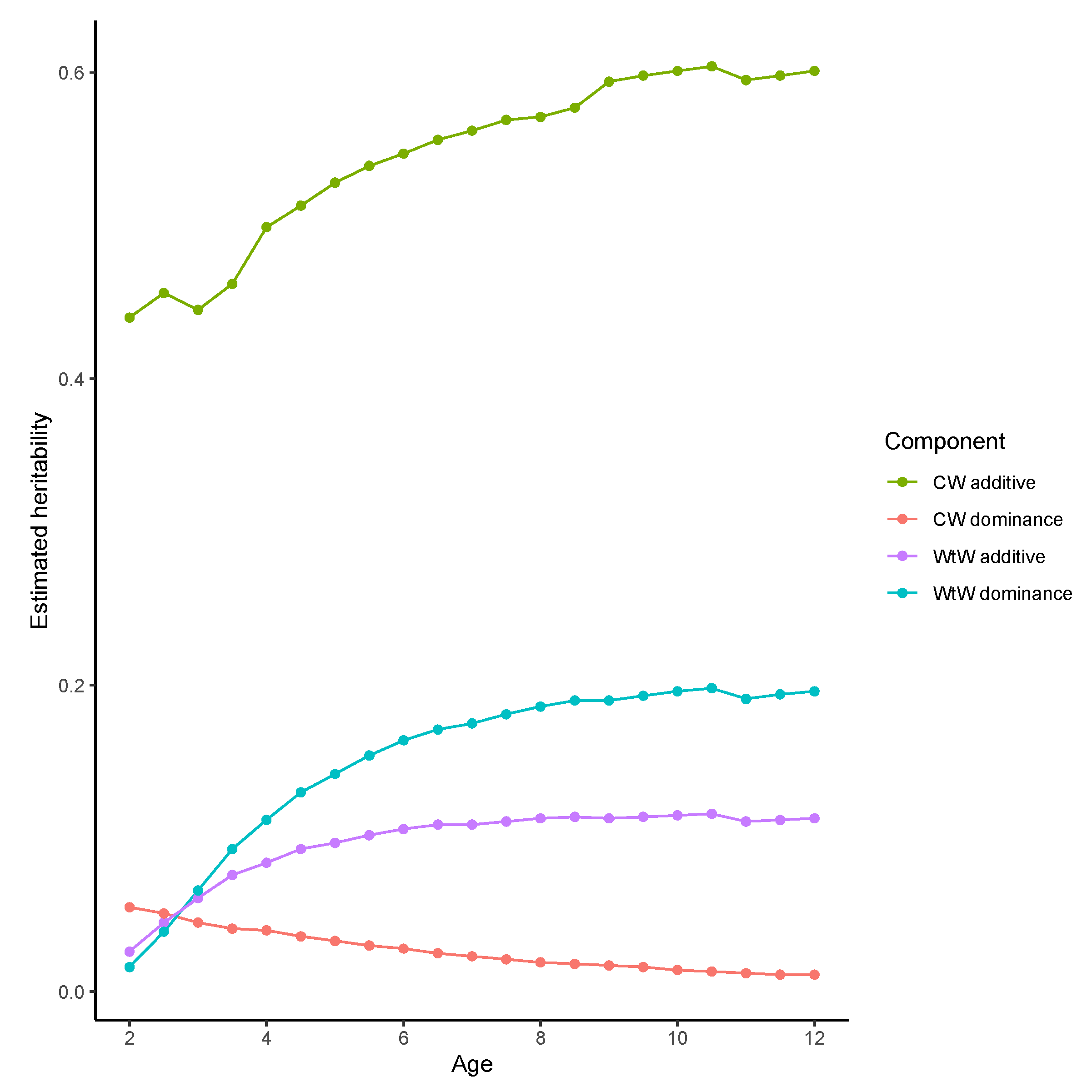
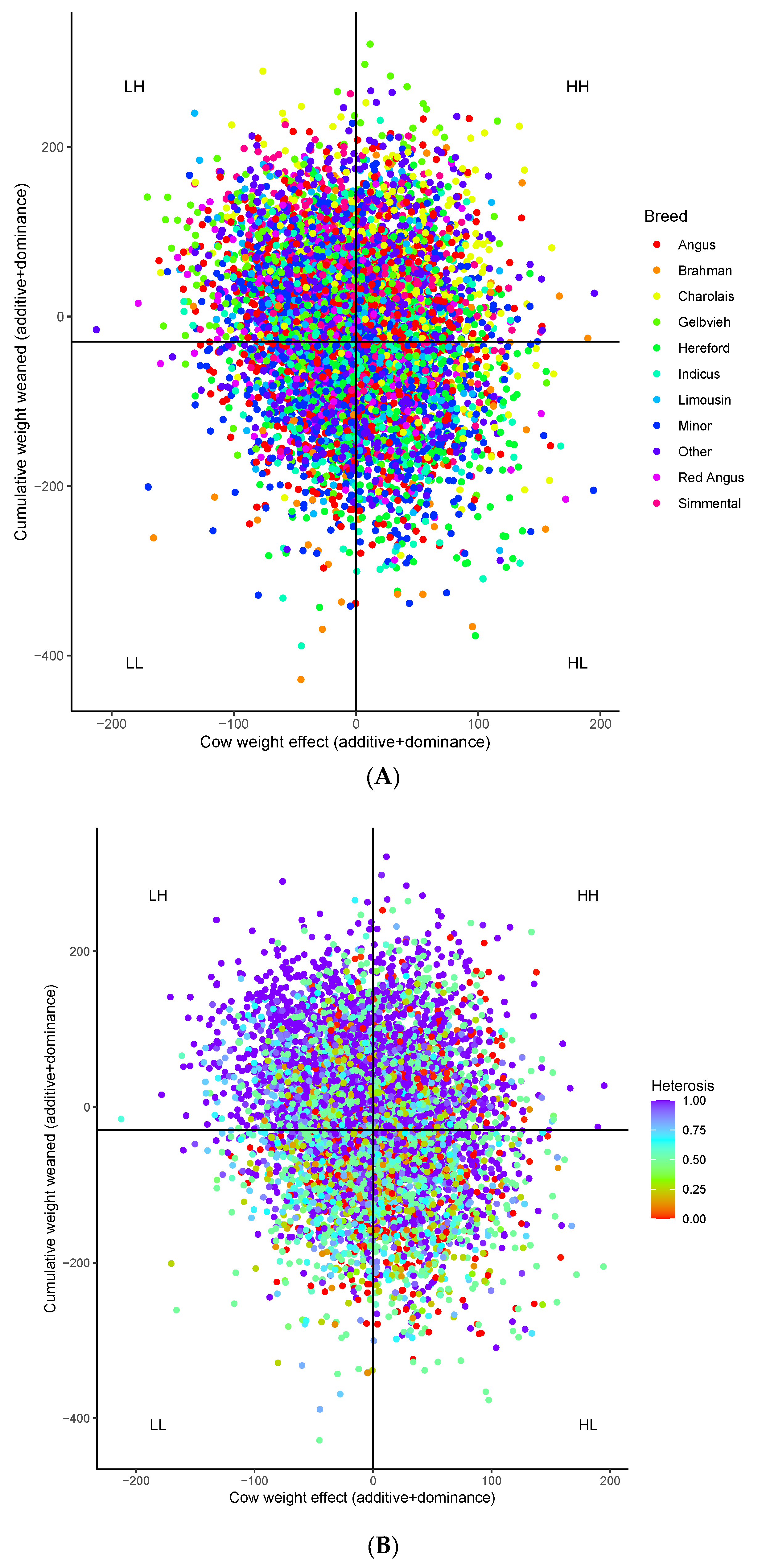
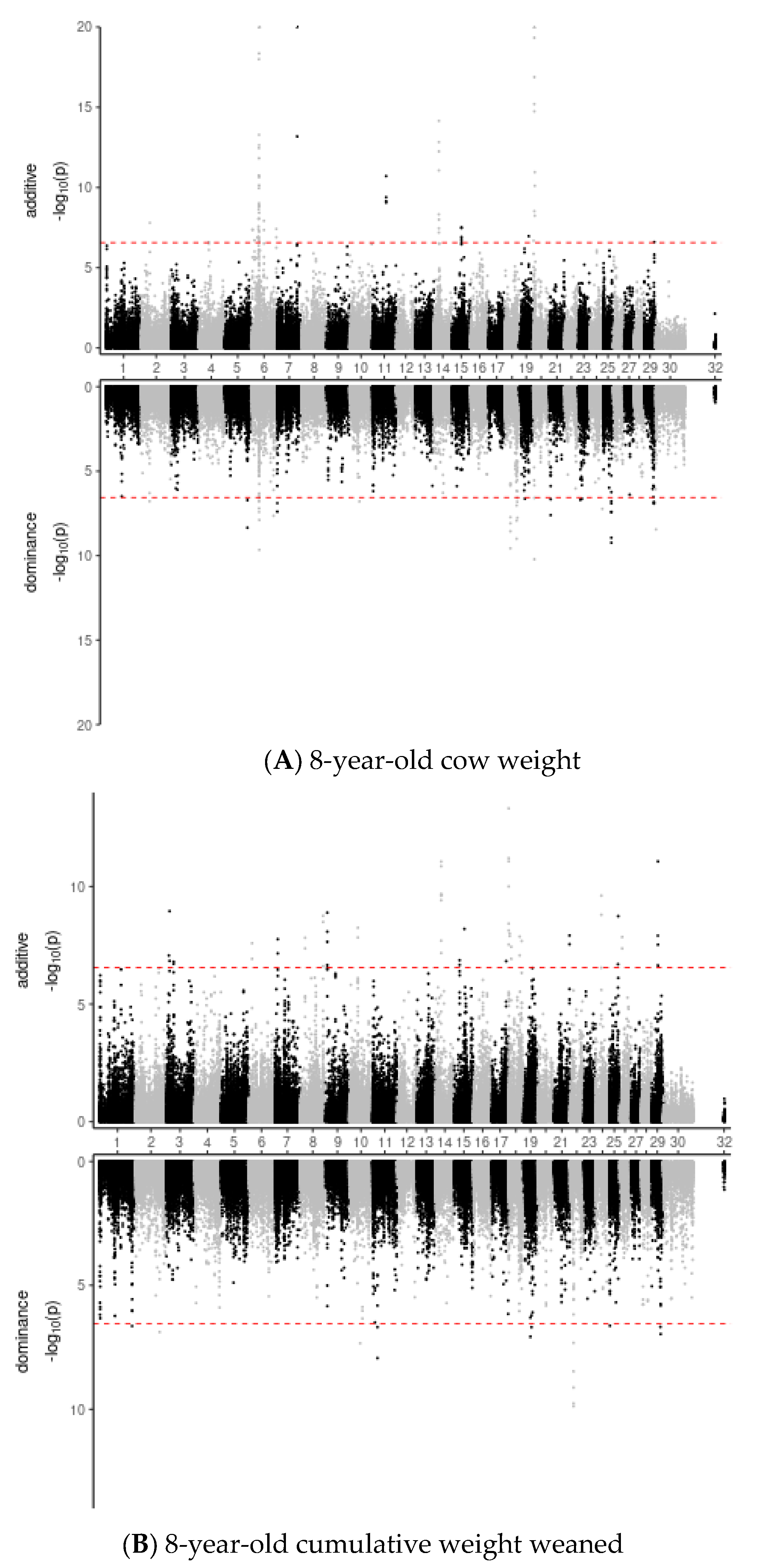
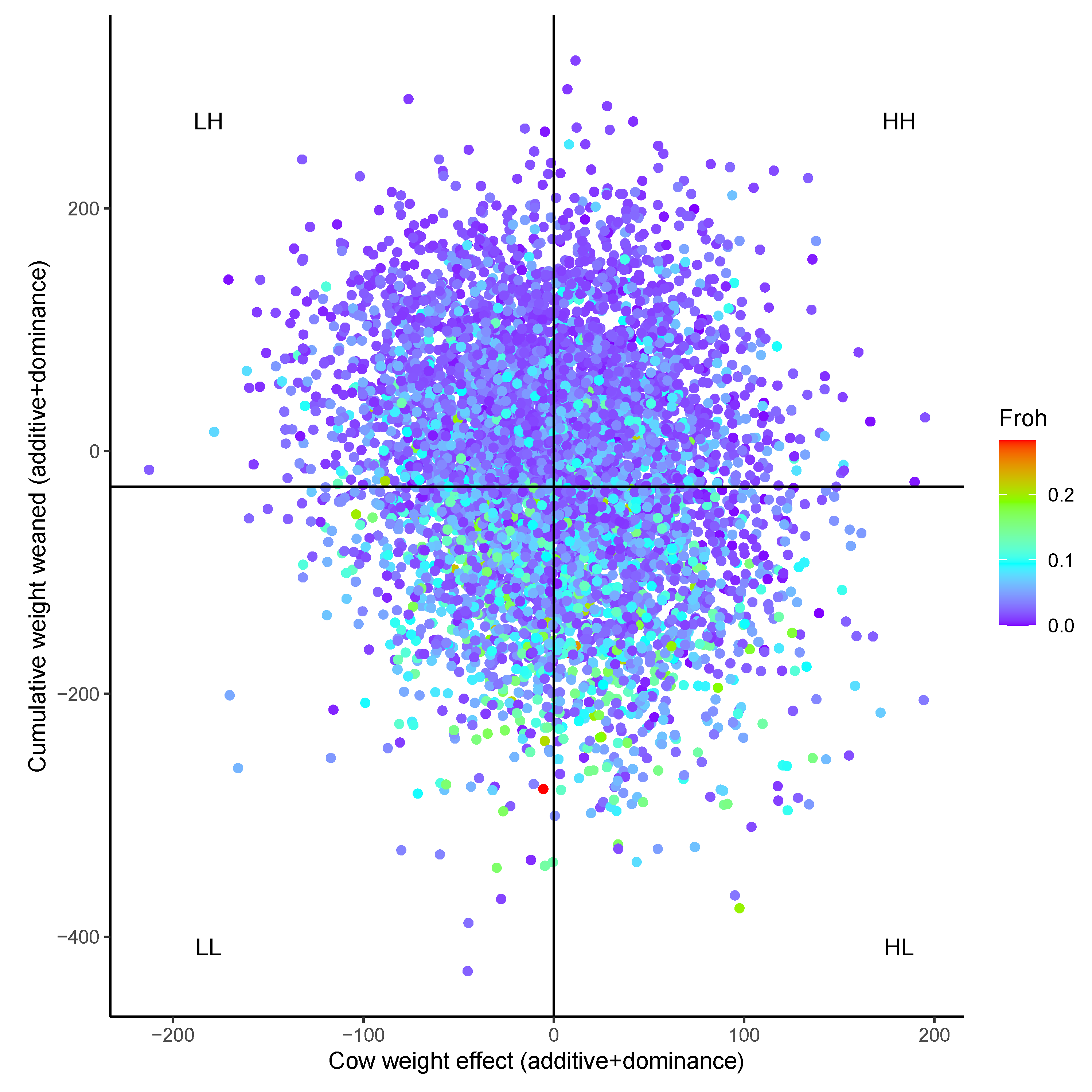
3. Results
3.1. Breed Contributions and Heterosis
3.2. Genomic Heterosis
3.3. Genomic Variant Effects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nature Conservancy. A Roadmap to a Sustainable Beef System A Collaborative Approach to Achieve Economic and Environmental Benefits for People and Nature; Nature Conservancy: Arlington County, VA, USA, 2020. [Google Scholar]
- Canadian Roundtable for Sustainable Beef. National Beef Sustainability Strategy; Canadian Roundtable for Sustainable Beef: Calgary, AB, Canada, 2016. [Google Scholar]
- US Roundtable for Sustainable Beef High-Priority Indicators. Available online: https://www.beefsustainability.us/high-priority-indicators (accessed on 30 August 2021).
- Global Roundtable of Sustainable Beef. GRSB Principles and Criteria for Defining Global Sustainable Beef; Global Roundtable for Sustainable Beef: Colorado Springs, CO, USA, 2017. [Google Scholar]
- Barbier, E.B. The Concept of Sustainable Economic Development. Environ. Conserv. 1987, 14, 101–110. [Google Scholar] [CrossRef]
- Golden, B.; Weerasinghe, S.; Crook, B.; Sanders, S.; Garrick, D. A Single-Step Hybrid Marker Effects Model Using Random Regression for Stayability in Hereford Cattle. In Proceedings of the World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018; Volume 296. [Google Scholar]
- USDA. Beef 2007–2008, Part IV: Reference of Beef Cow-Calf Management Practices in the United States; USDA, Animal and Plant Health Inspection Service, Veterinary Services, National Animal Health Monitoring System: Fort Collins, CO, USA, 2008.
- Proctor, L.E.; Archer, J.A.; Sise, J.A.; Zhang, X.; Crowley, J.J.; Amer, P.R. Impact of Trait Genetic Gains on Methane Emissions from NZ Beef and Dairy Farms. N. Z. J. Anim. Sci. Prod. 2020, 80, 76–79. [Google Scholar]
- Snelling, W.M.; Kuehn, L.A.; Thallman, R.M.; Bennett, G.L.; Golden, B.L. Genetic Correlations among Weight and Cumulative Productivity of Crossbred Beef Cows1. J. Anim. Sci. 2019, 97, 63–77. [Google Scholar] [CrossRef] [PubMed]
- National Academies of Sciences, Engineering, and Medicine (NASEM). Nutrient Requirements of Beef Cattle; NASEM: Washington, DC, USA, 2016. [Google Scholar]
- Johnson, K.A.; Johnson, D.E. Methane Emissions from Cattle. J. Anim. Sci. 1995, 73, 2483–2492. [Google Scholar] [CrossRef]
- Zimmermann, M.J.; Kuehn, L.A.; Spangler, M.L.; Thallman, R.M.; Snelling, W.M.; Lewis, R.M. Comparison of Different Functions to Describe Growth from Weaning to Maturity in Crossbred Beef Cattle1. J. Anim. Sci. 2019, 97, 1523–1533. [Google Scholar] [CrossRef]
- Henderson, B.B.; Gerber, P.J.; Hilinski, T.E.; Falcucci, A.; Ojima, D.S.; Salvatore, M.; Conant, R.T. Greenhouse Gas Mitigation Potential of the World’s Grazing Lands: Modeling Soil Carbon and Nitrogen Fluxes of Mitigation Practices. Agric. Ecosyst. Environ. 2015, 207, 91–100. [Google Scholar] [CrossRef]
- Adams, D.C.; Clark, R.T.; Klopfenstein, T.J.; Volesky, J.D. Nutritional Value of Grazed Forages and How It Fits The Cow’s Requirement Requirement. In Proceedings of the Range Beef Cow Symposium XIV, Gering, NE, USA, 5–7 December 1995. [Google Scholar]
- Li, J.H.; Mazur, C.A.; Berisa, T.; Pickrell, J.K. Low-Pass Sequencing Increases the Power of GWAS and Decreases Measurement Error of Polygenic Risk Scores Compared to Genotyping Arrays. Genome Res. 2021, 31, 529–537. [Google Scholar] [CrossRef]
- Snelling, W.M.; Hoff, J.L.; Li, J.H.; Kuehn, L.A.; Keel, B.N.; Lindholm-Perry, A.K.; Pickrell, J.K. Assessment of Imputation from Low-Pass Sequencing to Predict Merit of Beef Steers. Genes 2020, 11, 1312. [Google Scholar] [CrossRef]
- FASS. Guide for the Care and Use of Agricultural Animals in Research and Teaching, 3rd ed.; FASS: Champaign, IL, USA, 2010. [Google Scholar]
- Wheeler, T.L.; Cundiff, L.V.; Shackelford, S.D.; Koohmaraie, M. Characterization of Biological Types of Cattle (Cycle VII): Carcass, Yield, and Longissimus Palatability Traits. J. Anim. Sci. 2005, 83, 196–207. [Google Scholar]
- Wheeler, T.L.; Cundiff, L.V.; Shackelford, S.D.; Koohmaraie, M. Characterization of Biological Types of Cattle (Cycle VIII): Carcass, Yield, and Longissimus Palatability Traits. J. Anim. Sci. 2010, 88, 3070–3083. [Google Scholar] [CrossRef]
- Ahlberg, C.M.; Kuehn, L.A.; Thallman, R.M.; Kachman, S.D.; Snelling, W.M.; Spangler, M.L. Breed Effects and Genetic Parameter Estimates for Calving Difficulty and Birth Weight in a Multibreed Population. J. Anim. Sci. 2016, 94, 1857–1864. [Google Scholar] [CrossRef]
- Loimpute-Public. Available online: https://gitlab.com/gencove/loimpute-public (accessed on 25 October 2020).
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Aken, B.L.; Ayling, S.; Barrell, D.; Clarke, L.; Curwen, V.; Fairley, S.; Fernandez Banet, J.; Billis, K.; García Girón, C.; Hourlier, T.; et al. The Ensembl Gene Annotation System. Database 2016, 2016, baw093. [Google Scholar] [CrossRef]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De Novo Assembly of the Cattle Reference Genome with Single-Molecule Sequencing. Gigascience 2020, 9, giaa021. [Google Scholar] [CrossRef]
- VanRaden, P.M.; Null, D.J.; Sargolzaei, M.; Wiggans, G.R.; Tooker, M.E.; Cole, J.B.; Sonstegard, T.S.; Connor, E.E.; Winters, M.; van Kaam, J.; et al. Genomic Imputation and Evaluation Using High-Density Holstein Genotypes. J. Dairy Sci. 2013, 96, 668–678. [Google Scholar] [CrossRef]
- Schnabel, R. ARS-UCD1.2 Cow Genome Assembly: Mapping of All Existing Variants. Available online: https://www.animalgenome.org/repository/cattle/UMC_bovine_coordinates/ (accessed on 3 October 2021).
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A High-Performance Computing Toolset for Relatedness and Principal Component Analysis of SNP Data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Su, G.; Christensen, O.F.; Ostersen, T.; Henryon, M.; Lund, M.S. Estimating Additive and Non-Additive Genetic Variances and Predicting Genetic Merits Using Genome-Wide Dense Single Nucleotide Polymorphism Markers. PLoS ONE 2012, 7, e45293. [Google Scholar] [CrossRef]
- Meyer, K. WOMBAT—A Tool for Mixed Model Analyses in Quantitative Genetics by Restricted Maximum Likelihood (REML). J. Zhejiang Univ. Sci. B 2007, 8, 815–821. [Google Scholar] [CrossRef]
- Dickerson, G.E. Inbreeding and Heterosis in Animals. J. Anim. Sci. 1973, 1973, 54–77. [Google Scholar] [CrossRef]
- Strandén, I.; Garrick, D.J. Technical Note: Derivation of Equivalent Computing Algorithms for Genomic Predictions and Reliabilities of Animal Merit. J. Dairy Sci. 2009, 92, 2971–2975. [Google Scholar] [CrossRef] [PubMed]
- Snelling, W.M.; Kachman, S.D.; Bennett, G.L.; Spangler, M.L.; Kuehn, L.A.; Thallman, R.M. Functional SNP Associated with Birth Weight in Independent Populations Identified with a Permutation Step Added to GBLUP-GWAS. J. Anim. Sci. 2017, 95, 97–98. [Google Scholar] [CrossRef][Green Version]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Building a Livestock Genetic and Genomic Information Knowledgebase through Integrative Developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2019, 47, D701–D710. [Google Scholar] [CrossRef] [PubMed]
- Marras, G.; Wood, B.J.; Makanjuola, B.; Malchiodi, F.; Peeters, K.; van As, P.; Baes, C.F.; Biscarini, F. Characterization of Runs of Homozygosity and Heterozygosity-Rich Regions in a Commercial Turkey (Meleagris Gallopavo) Population. In Proceedings of the World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018; Species–Avian 1. p. 763. [Google Scholar]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. DetectRUNS: Vignettes/DetectRUNS.Vignette.Rmd. Available online: https://rdrr.io/cran/detectRUNS/f/vignettes/detectRUNS.vignette.Rmd (accessed on 16 May 2022).
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P.P. Analysis of Runs of Homozygosity and Their Relationship with Inbreeding in Five Cattle Breeds Farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Kimchi-Sarfaty, C. Understanding the Contribution of Synonymous Mutations to Human Disease. Nat. Rev. Genet. 2011, 12, 683–691. [Google Scholar] [CrossRef]
- Hunt, R.C.; Simhadri, V.L.; Iandoli, M.; Sauna, Z.E.; Kimchi-Sarfaty, C. Exposing Synonymous Mutations. Trends Genet. 2014, 30, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Phocas, F.; Bloch, C.; Chapelle, P.; Bécherel, F.; Renand, G.; Ménissier, F. Developing a Breeding Objective for a French Purebred Beef Cattle Selection Programme. Livest. Prod. Sci. 1998, 57, 49–65. [Google Scholar] [CrossRef]
- Newman, S.; Morris, C.A.; Baker, R.L.; Nicoll, G.B. Genetic Improvement of Beef Cattle in New Zealand: Breeding Objectives. Livest. Prod. Sci. 1992, 32, 111–130. [Google Scholar] [CrossRef]
- MacNeil, M.D.; Newman, S.; Enns, R.M.; Stewart-Smith, J. Relative Economic Values for Canadian Beef Production Using Specialized Sire and Dam Lines. Can. J. Anim. Sci. 1994, 74, 411–417. [Google Scholar] [CrossRef]
- Tang, G.; Stewart-Smith, J.; Plastow, G.; Moore, S.; Basarab, J.; MacNeil, M.; Wang, Z. Optimizing a Beef Production System Using Specialized Sire and Dam Lines. Can. J. Anim. Sci. 2011, 91, 353–361. [Google Scholar] [CrossRef]
- Gregory, K.E.; Cundiff, L.V.; Koch, R.M. Breed Effects and Heterosis in Advanced Generations of Composite Populations for Preweaning Traits of Beef Cattle. J. Anim. Sci. 1991, 69, 947. [Google Scholar] [CrossRef]
- Spangler, M.L. The Value of Heterosis in Cow Herds: Lessons From the Past That Apply to Today. In Proceedings of the Range Beef Cow Symposium XX, Fort Collins, CO, USA, 11–13 December 2007. [Google Scholar]
- Sumreddee, P.; Toghiani, S.; Hay, E.H.; Roberts, A.; Agrrey, S.E.; Rekaya, R. Inbreeding Depression in Line 1 Hereford Cattle Population Using Pedigree and Genomic Information1. J. Anim. Sci. 2019, 97, 1–18. [Google Scholar] [CrossRef]
- Doekes, H.P.; Veerkamp, R.F.; Bijma, P.; de Jong, G.; Hiemstra, S.J.; Windig, J.J. Inbreeding Depression Due to Recent and Ancient Inbreeding in Dutch Holstein–Friesian Dairy Cattle. Genet. Sel. Evol. 2019, 51, 54. [Google Scholar] [CrossRef]
- Jonker, A.; Green, P.; Waghorn, G.; van der Weerden, T.; Pacheco, D.; de Klein, C. A Meta-Analysis Comparing Four Measurement Methods to Determine the Relationship between Methane Emissions and Dry-Matter Intake in New Zealand Dairy Cattle. Anim. Prod. Sci. 2020, 60, 96–101. [Google Scholar] [CrossRef]
- Greenwood, P.L.; Paull, D.R.; McNally, J.; Kalinowski, T.; Ebert, D.; Little, B.; Smith, D.v.; Rahman, A.; Valencia, P.; Ingham, A.B.; et al. Use of Sensor-Determined Behaviours to Develop Algorithms for Pasture Intake by Individual Grazing Cattle. Crop Pasture Sci. 2017, 68, 1091–1099. [Google Scholar] [CrossRef]
- Cottle, D.J. Optimising Natural 13C Marker Based Pasture Intake Estimates for Cattle Using a Genetic Algorithm Approach. Livest. Sci. 2017, 197, 53–60. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | SNP | Sires | Dams | Nonparents | Total |
|---|---|---|---|---|---|
| SNP50 v1 1 | 54,115 | 1245 | 1064 | 2466 | 4775 |
| SNP50 v2 1 | 54,042 | 90 | 956 | 4140 | 5186 |
| BovineHD 1 | 774,990 | 921 | 467 | 162 | 1550 |
| GGP 2-F250 3 | 206,629 | 1435 | 561 | 371 | 2367 |
| GGP v1 2 | 76,570 | 0 | 0 | 517 | 517 |
| GGP v2 2 | 19,640 | 0 | 0 | 172 | 172 |
| GGP v3 2 | 25,969 | 0 | 816 | 2635 | 3451 |
| GGP v4 2 | 29,704 | 0 | 154 | 789 | 943 |
| GGP 50 K 2 | 44,739 | 0 | 1210 | 2612 | 3822 |
| GGP 100 K 2 | 93,843 | 1 | 177 | 971 | 1149 |
| All arrays | 911,640 | 1886 | 4917 | 14,567 | 21,370 |
| Low-pass | 59,204,180 | 412 | 2375 | 136 | 2923 |
| Low-pass + arrays | 59,280,638 | 2013 | 6088 | 14,675 | 22,776 |
| Trait | Minimum | Maximum | Mean | SD | |
|---|---|---|---|---|---|
| CW | observed | 284.4 | 947.8 | 609.1 | 82.5 |
| additive | −197.5 | 194.3 | 0.4 | 50.8 | |
| dominance | −15.1 | 20.3 | −0.5 | 3.7 | |
| WtW | observed | 0.0 | 1955.6 | 1286.3 | 253.8 |
| additive | −297.6 | 163.2 | −3.4 | 55.3 | |
| dominance | −267.6 | 289.2 | −10.0 | 65.4 |
| Dominance | Total | |||
|---|---|---|---|---|
| CW | WtW | CW | WtW | |
| pHet | 6.53 | 13.14 | 1.52 | 5.61 |
| gHet | 17.63 | 11.08 | 0.46 | 0.03 |
| Fg | 6.95 | 1.22 | 1.02 | 3.40 |
| HRR | 4.12 | 0.90 | 0.57 | 0.38 |
| ROH | 12.60 | 21.26 | 0.01 | 11.65 |
| Cow Group 2 | Genomic Indicators | |||||
|---|---|---|---|---|---|---|
| pHet | gHet | Fg | HRR | ROH | n | |
| All | 0.711 (0.004) | 0.349 (0.0003) | 0.006 (0.002) | 9072758 | 106867400 | 6211 |
| (26512) | (1193523) | |||||
| Low CW | 0.742 (0.005) | 0.347 (0.0005) | −0.006 (0.002) | 9072758 | 106584500 | 3114 |
| (34543) | (1662454) | |||||
| High CW | 0.680 (0.006) | 0.350 (0.0005) | 0.018 (0.003) | 9182985 | 107151900 | 3097 |
| (40165) | (1713475) | |||||
| Low WtW | 0.700 (0.006) | 0.353 (0.0006) | 0.036 (0.003) | 9389333 | 117693600 | 3065 |
| (40696) | (1820098) | |||||
| High WtW | 0.769 (0.005) | 0.349 (0.0004) | −0.011 (0.002) | 8961127 | 81655680 | 3219 |
| (35280) | (1220320) | |||||
| LL | 0.662 (0.009) | 0.347 (0.0008) | 0.016 (0.004) | 8992696 | 137128100 | 1356 |
| (53574) | (2934484) | |||||
| LH | 0.803 (0.006) | 0.347 (0.0005) | −0.022 (0.002) | 8940328 | 83025260 | 1758 |
| (45131) | (1681042) | |||||
| HH | 0.727 (0.009) | 0.352 (0.0007) | 0.003 (0.003) | 8986154 | 80007690 | 1461 |
| (55628) | (1770986) | |||||
| HL | 0.637 (0.008) | 0.349 (0.0009) | 0.031 (0.004) | 9358762 | 131392600 | 1636 |
| (57228) | (2694706) | |||||
| CW | WtW | |||||
|---|---|---|---|---|---|---|
| All | Additive | Dominance | Additive | Dominance | ||
| Variants Genes | 366 | 139 | 68 | 120 | 46 | |
| 120 | 46 | 27 | 37 | 15 | ||
| Functional annotation | ||||||
| Impact | Annotation | |||||
| HIGH | splice donor; intron | 1 | 1 | |||
| HIGH | start_lost | 1 | 1 | |||
| HIGH | stop_gained | 1 | 1 | |||
| MODERATE | nonsynonymous | 67 | 31 | 7 | 26 | 5 |
| MODIFIER | 3′ UTR | 111 | 45 | 31 | 20 | 16 |
| MODIFIER | 5′ UTR | 33 | 17 | 4 | 10 | 3 |
| MODIFIER | noncoding exon | 16 | 3 | 9 | 4 | |
| LOW | 5′ UTR; premature start codon gain | 1 | 1 | 1 | ||
| LOW | splice region; intron | 11 | 3 | 2 | 4 | 2 |
| LOW | splice region; synonymous | 2 | 1 | 1 | ||
| LOW | synonymous | 131 | 39 | 23 | 54 | 17 |
| CW | WtW | ||||
|---|---|---|---|---|---|
| Trait Category | All | Additive | Dominance | Additive | Dominance |
| Exterior | 79 (8.0) | 29 (5.4) | 36 (11.4) | 33 (11.0) | 15 (10.1) |
| Health | 91 (9.2) | 35 (6.5) | 44 (13.9) | 36 (12.0) | 17 (11.4) |
| Meat and Carcass | 238 (24.1) | 150 (27.9) | 60 (18.9) | 67 (22.3) | 28 (18.8) |
| Milk | 171 (17.3) | 92 (17.1) | 78 (24.6) | 58 (19.3) | 38 (25.5) |
| Production | 313 (31.7) | 211 (39.3) | 61 (19.2) | 60 (20.0) | 33 (22.1) |
| Reproduction | 94 (9.5) | 20 (3.7) | 38 (12.0) | 46 (15.3) | 18 (12.1) |
| Total | 986 (100) | 537 (100) | 317 (100) | 300 (100) | 149 (100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snelling, W.M.; Thallman, R.M.; Spangler, M.L.; Kuehn, L.A. Breeding Sustainable Beef Cows: Reducing Weight and Increasing Productivity. Animals 2022, 12, 1745. https://doi.org/10.3390/ani12141745
Snelling WM, Thallman RM, Spangler ML, Kuehn LA. Breeding Sustainable Beef Cows: Reducing Weight and Increasing Productivity. Animals. 2022; 12(14):1745. https://doi.org/10.3390/ani12141745
Chicago/Turabian StyleSnelling, Warren M., R. Mark Thallman, Matthew L. Spangler, and Larry A. Kuehn. 2022. "Breeding Sustainable Beef Cows: Reducing Weight and Increasing Productivity" Animals 12, no. 14: 1745. https://doi.org/10.3390/ani12141745
APA StyleSnelling, W. M., Thallman, R. M., Spangler, M. L., & Kuehn, L. A. (2022). Breeding Sustainable Beef Cows: Reducing Weight and Increasing Productivity. Animals, 12(14), 1745. https://doi.org/10.3390/ani12141745