
Comparative Analysis for Genetic Characterization in Korean Native Jeju Horse

 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and DNA Extraction
2.2. Whole-Genome Resequencing (WGRS)
2.3. Data Analysis
2.4. PCR and DNA Sequencing Analysis
2.5. Sliding-Window Analysis of dN/dS (Positive Selection Analysis)
2.6. Genotyping Assay Using Digital PCR
3. Results
3.1. Sequencing, Read Mapping, and Genomic Variant Detection
3.2. Functional Annotation of Nonsynonymous
3.3. Positive Selection of eqCD1a6 Gene in Jeju Horses
3.4. Genotyping Assay for Molecular Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ludwig, A.; Pruvost, M.; Reissmann, M.; Benecke, N.; Brockmann, G.A.; Castanos, P.; Cieslak, M.; Lippold, S.; Llorente, L.; Malaspinas, A.S.; et al. Coat color variation at the beginning of horse domestication. Science 2009, 324, 485. [Google Scholar] [CrossRef] [PubMed]
- Outram, A.K.; Stear, N.A.; Bendrey, R.; Olsen, S.; Kasparov, A.; Zaibert, V.; Thorpe, N.; Evershed, R.P. The earliest horse harnessing and milking. Science 2009, 323, 1332–1335. [Google Scholar] [CrossRef] [PubMed]
- Lippold, S.; Matzke, N.J.; Reissmann, M.; Hofreiter, M. Whole mitochondrial genome sequencing of domestic horses reveals incorporation of extensive wild horse diversity during domestication. BMC Evol. Biol. 2011, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.L.; Mickelson, J.R.; Rendahl, A.K.; Valberg, S.J.; Andersson, L.S.; Axelsson, J.; Bailey, E.; Bannasch, D.; Binns, M.M.; Borges, A.S.; et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, Q.; Sun, J.; Li, X.; Xu, L.; Yang, H.; Shi, H.; Ning, S.; Chen, L.; Li, Y.; et al. In silico detection and characteristics of novel microRNA genes in the Equus caballus genome using an integrated ab initio and comparative genomic approach. Genomics 2009, 94, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Gim, J.A.; Hong, C.P.; Kim, D.S.; Moon, J.W.; Choi, Y.; Eo, J.; Kwon, Y.J.; Lee, J.R.; Jung, Y.D.; Bae, J.H.; et al. Genome-wide analysis of DNA methylation before-and after exercise in the thoroughbred horse with MeDIP-Seq. Mol. Cells 2015, 38, 210–220. [Google Scholar] [CrossRef]
- Kim, N.Y.; Seong, H.S.; Kim, D.C.; Park, N.G.; Yang, B.C.; Son, J.K.; Shin, S.M.; Woo, J.H.; Shin, M.C.; Yoo, J.H.; et al. Genome-wide analyses of the Jeju, Thoroughbred, and Jeju crossbred horse populations using the high density SNP array. Genes Genom. 2018, 40, 1249–1258. [Google Scholar] [CrossRef]
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef]
- Park, K.D.; Park, J.; Ko, J.; Kim, B.C.; Kim, H.S.; Ahn, K.; Do, K.T.; Choi, H.; Kim, H.M.; Song, S.; et al. Whole transcriptome analyses of six thoroughbred horses before and after exercise using RNA-Seq. BMC Genom. 2012, 13, 473. [Google Scholar] [CrossRef]
- Yang, Y.H.; Kim, K.I.; Cothran, E.G.; Flannery, A.R. Genetic diversity of Cheju horses (Equus caballus) determined by using mitochondrial DNA D-loop polymorphism. Biochem. Genet. 2002, 40, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ni, P.; Ahmad, H.I.; Gemingguli, M.; Baizilaitibei, A.; Gulibaheti, D.; Fang, Y.; Wang, H.; Asif, A.R.; Xiao, C.; et al. Detecting the Population Structure and Scanning for Signatures of Selection in Horses (Equus caballus) from Whole-Genome Sequencing Data. Evol. Bioinform. Online 2018, 14, 1176934318775106. [Google Scholar] [CrossRef]
- Do, K.T.; Lee, J.H.; Lee, H.K.; Kim, J.; Park, K.D. Estimation of effective population size using single-nucleotide polymorphism (SNP) data in Jeju horse. J. Anim. Sci. Technol. 2014, 56, 28. [Google Scholar] [CrossRef]
- Yoon, S.H.; Kim, J.; Shin, D.; Cho, S.; Kwak, W.; Lee, H.K.; Park, K.D.; Kim, H. Complete mitochondrial genome sequences of Korean native horse from Jeju Island: Uncovering the spatio-temporal dynamics. Mol. Biol. Rep. 2017, 44, 233–242. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Parmley, J.L.; Hurst, L.D. How common are intragene windows with KA > KS owing to purifying selection on synonymous mutations? J. Mol. Evol. 2007, 64, 646–655. [Google Scholar] [CrossRef]
- Metzger, J.; Tonda, R.; Beltran, S.; Agueda, L.; Gut, M.; Distl, O. Next generation sequencing gives an insight into the characteristics of highly selected breeds versus non-breed horses in the course of domestication. BMC Genom. 2014, 15, 562. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Porcelli, S.A.; Modlin, R.L. The CD1 system: Antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu. Rev. Immunol. 1999, 17, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Herron, L.L.; Kapur, V.; Meijer, W.G.; Byrne, B.A.; Ren, J.; Nicholson, V.M.; Prescott, J.F. Partial genome sequencing of Rhodococcus equi ATCC 33701. Vet. Microbiol. 2003, 94, 143–158. [Google Scholar] [CrossRef]
- Park, S.H.; Bendelac, A. CD1-restricted T-cell responses and microbial infection. Nature 2000, 406, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Dossa, R.G.; Alperin, D.C.; Hines, M.T.; Hines, S.A. The equine CD1 gene family is the largest and most diverse yet identified. Immunogenetics 2014, 66, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Proutski, V.; Holmes, E. SWAN: Sliding window analysis of nucleotide sequence variability. Bioinformatics 1998, 14, 467–468. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sommer, S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2005, 2, 16. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Jeju_1 | Jeju_2 | Jeju_3 | Jeju_4 | Jeju_5 | Thoroughbred | Average |
|---|---|---|---|---|---|---|---|
| Total reads a | 690,508,306 | 609,871,934 | 661,791,072 | 617,836,736 | 606,111,944 | 990,419,468 | 696,089,910 |
| Clean reads | 621,253,885 | 532,985,097 | 581,568,986 | 555,881,352 | 545,197,568 | 820,451,365 | 609,556,376 |
| Clean reads, % b | 89.97% | 87.39% | 87.88% | 89.97% | 89.95% | 82.84% | 88% |
| Mapped reads | 612,618,456 | 523,657,858 | 572,263,882 | 547,987,837 | 536,038,249 | 780,311,183 | 595,479,578 |
| Mapped reads, % c | 98.61% | 98.25% | 98.40% | 98.58% | 98.32% | 95.11% | 97.88% |
| Average Depth | 37.24x | 31.48x | 34.50x | 33.14x | 32.36x | 46.01x | 35.79x |
| Type a | Jeju_1 | Jeju_2 | Jeju_3 | Jeju_4 | Jeju_5 | Thoroughbred |
|---|---|---|---|---|---|---|
| Homo INS | 246,788 | 235,661 | 242,743 | 238,128 | 239,981 | 157,841 |
| Hetero INS | 199,722 | 187,896 | 193,519 | 201,288 | 195,073 | 142,173 |
| Total INS | 446,510 | 423,557 | 436,262 | 439,416 | 435,054 | 300,014 |
| Homo DEL | 263,896 | 254,611 | 259,821 | 255,380 | 258,247 | 166,728 |
| Hetero DEL | 201,114 | 190,505 | 196,679 | 204,404 | 196,586 | 122,802 |
| Total DEL | 465,010 | 445,116 | 456,500 | 459,784 | 454,833 | 289,530 |
| Homo SNP | 2,404,789 | 2,331,653 | 2,415,831 | 2,329,651 | 2,369,765 | 1,740,637 |
| Hetero SNP | 4,304,466 | 4,211,637 | 4,312,941 | 4,397,561 | 4,355,094 | 2,202,227 |
| Total SNP | 6,709,255 | 6,543,290 | 6,728,772 | 6,727,212 | 6,724,859 | 3,942,864 |
| Region | SNP | INS | DEL |
|---|---|---|---|
| Upstream | 7715 | 10,497 | 7336 |
| Exon | 1138 | 1484 | 903 |
| Intergenic | 63,497 | 84,897 | 46,518 |
| intragenic | 119 | 190 | 112 |
| Intron | 21,312 | 37,272 | 19,557 |
| Splice site | 194 | 595 | 435 |
| 5’ UTR | 174 | 307 | 197 |
| 3’ UTR | 43 | 106 | 57 |
| Total | 94,192 | 135,348 | 75,115 |
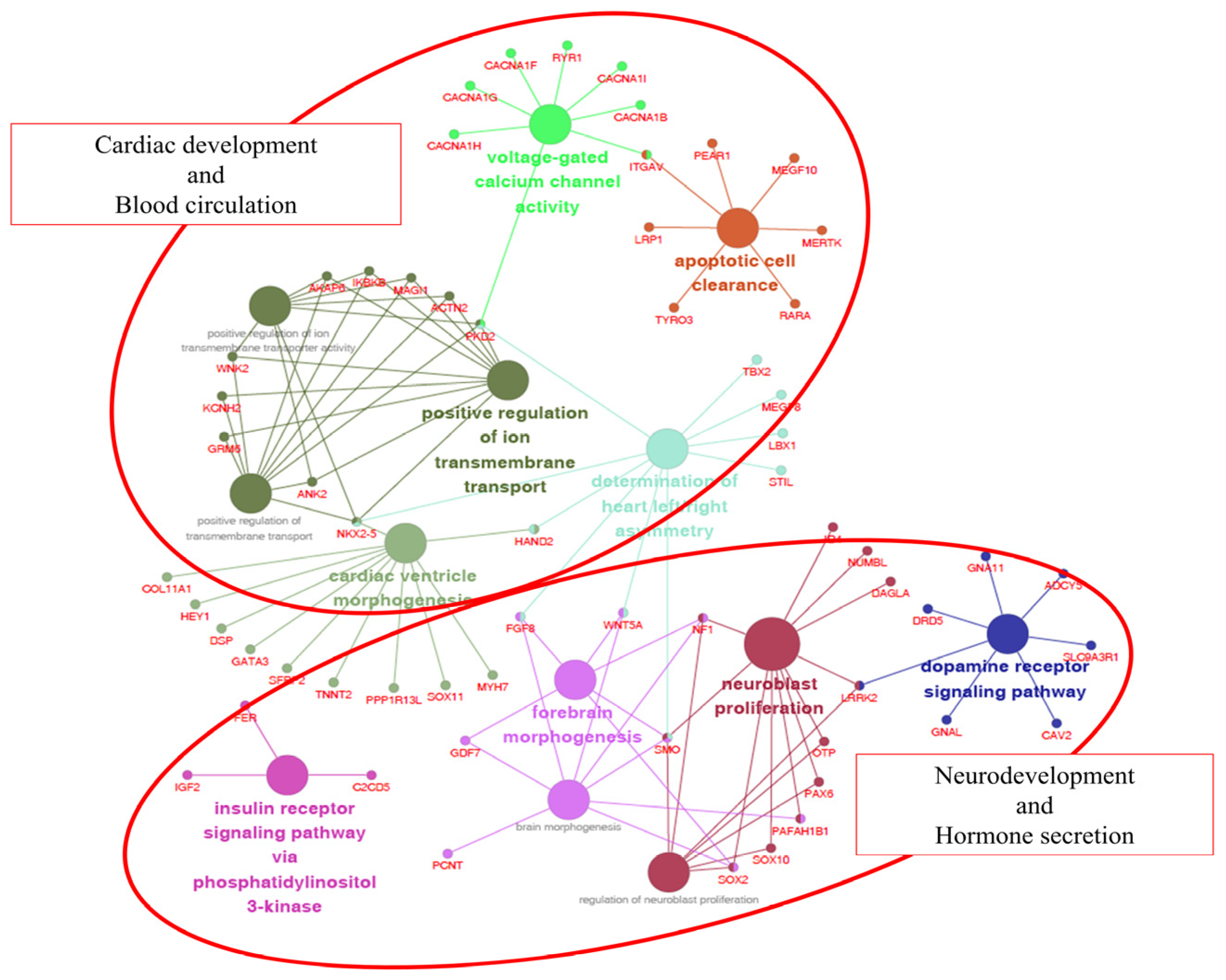
| Gene Ontology | Count | Gene Symbol | p-Value | Adjusted p-Value |
|---|---|---|---|---|
| GO:0003208 Cardiac ventricle morphogenesis | 11 | COL11A1, DSP, GATA3, HAND2, HEY1, MYH7, NKX2-5, PPP1R13L, SFRP2, SOX11, TNNT2 | 0.000560 | 0.032501 |
| GO:0005245 Voltage-gated calcium channel activity | 8 | CACNA1B, CACNA1F, CACNA1G, CACNA1H, CACNA1I, ITGAV, PKD2, RYR1 | 0.000835 | 0.046773 |
| GO:0061371 Determination of heart left/right asymmetry | 10 | FGF8, HAND2, LBX1, MEGF8, NKX2-5, PKD2, SMO, STIL, TBX2, WNT5A | 0.000593 | 0.033776 |
| GO:0007212 Dopamine receptor signaling pathway | 7 | ADCY5, CAV2, DRD5, GNA11, GNAL, LRRK2, SLC9A3R1 | 0.000292 | 0.018073 |
| GO:0038028 Insulin receptor signaling pathway via phosphatidylinositol 3-kinase | 3 | C2CD5, FER, IGF2 | 0.000252 | 0.016145 |
| GO:0043277 Apoptotic cell clearance | 7 | ITGAV, LRP1, MEGF10, MERTK, PEAR1, RARA, TYRO3 | 0.000210 | 0.013660 |
| GO:0048853 Forebrain morphogenesis | 6 | FGF8, GDF7, NF1, SMO, SOX2, WNT5A | 0.000426 | 0.025571 |
| GO:0048854 Brain morphogenesis | 8 | FGF8, GDF7, NF1, PAFAH1B1, PCNT, SMO, SOX2, WNT5A | 0.000531 | 0.031340 |
| GO:0007405 Neuroblast proliferation | 11 | DAGLA, ID4, LRRK2, NF1, NUMBL, OTP, PAFAH1B1, PAX6, SMO, SOX10, SOX2 | 0.000068 | 0.004559 |
| GO:1902692 Regulation of neuroblast proliferation | 7 | LRRK2, NF1, OTP, PAX6, SMO, SOX10, SOX2 | 0.000900 | 0.049499 |
| GO:0034764 Positive regulation of transmembrane transport | 10 | ACTN2, AKAP6, ANK2, GRM6, IKBKB, KCNH2, MAGI1, NKX2-5, PKD2, WNK2 | 0.000276 | 0.017401 |
| GO:0034767 Positive regulation of ion transmembrane transport | 10 | ACTN2, AKAP6, ANK2, GRM6, IKBKB, KCNH2, MAGI1, NKX2-5, PKD2, WNK2 | 0.000181 | 0.011963 |
| GO:0032414 Positive regulation of ion transmembrane transporter activity | 8 | ACTN2, AKAP6, ANK2, IKBKB, MAGI1, NKX2-5, PKD2, WNK2 | 0.000324 | 0.019737 |
| Gene | Classification | No. of SNP |
|---|---|---|
| SNPs | 275 | |
| Upstream gene variant | 162 | |
| Exon variant | 51 | |
| Intron variant | 62 | |
| eqCD1a6 | INDELs | 21 |
| Upstream gene variant | 19 | |
| Intron variant | 2 | |
| Total | 296 |
| Protein | Position | AA1 | AA2 | Description | Prediction Score |
|---|---|---|---|---|---|
| eqCD1a6 | 6 (Exon 1) | F | L | Unknown | - |
| 33 (Exon 2) | S | I | Benign | 0.000 | |
| 39 (Exon 2) | P | R | Benign | 0.000 | |
| 41 (Exon 2) | R | W | Benign | 0.000 | |
| 60 (Exon 2) | N | S | Benign | 0.001 | |
| 64 (Exon 2) | S | T | Benign | 0.002 | |
| 65 (Exon 2) | F | L | Benign | 0.128 | |
| 67 (Exon 2) | F | Y | Benign | 0.156 | |
| 78 (Exon 2) | S | N | Benign | 0.000 | |
| 79 (Exon 2) | E | K | Benign | 0.127 | |
| 81 (Exon 2) | L | M | Benign | 0.311 | |
| 84 (Exon 2) | I | M | Benign | 0.007 | |
| 88 (Exon 2) | F | S | Benign | 0.452 | |
| 98 (Exon 2) | I | V | Benign | 0.000 | |
| 109 (Exon 2) | A | E | Benign | 0.000 | |
| 113 (Exon 3) | Q | H | Possibly damaging | 0.871 | |
| 116 (Exon 3) | L | V | Benign | 0.003 | |
| 117 (Exon 3) | A | T | Benign | 0.009 | |
| 118 (Exon 3) | M | K | Benign | 0.001 | |
| 132 (Exon 3) | A | M | Benign | 0.002 | |
| 139 (Exon 3) | S | L | Benign | 0.311 | |
| 142 (Exon 3) | I | A | Benign | 0.401 | |
| 144 (Exon 3) | I | F | Benign | 0.001 | |
| 157 (Exon 3) | S | R | Benign | 0.044 | |
| 164 (Exon 3) | G | R | Benign | 0.000 | |
| 165 (Exon 3) | R | L | Benign | 0.000 | |
| 171 (Exon 3) | V | I | Benign | 0.021 | |
| 172 (Exon 3) | F | I | Benign | 0.001 | |
| 174 (Exon 3) | E | G | Possibly damaging | 0.950 | |
| 175 (Exon 3) | T | I | Benign | 0.001 | |
| 176 (Exon 3) | W | T | Benign | 0.001 | |
| 219 (Exon 4) | H | R | Benign | 0.001 | |
| 225 (Exon 4) | H | Q | Probably damaging | 1.000 | |
| 228 (Exon 4) | S | G | Benign | 0.000 | |
| 242 (Exon 4) | K | E | Benign | 0.000 | |
| 243 (Exon 4) | K | Q | Benign | 0.001 | |
| 301 (Exon 5) | T | M | Benign | 0.016 |
| Species | Sample | Total Well | Valid Well | Ch01(FAM) | Ch02(Cy5) | Ratio (Ch01/Ch02) | Result | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Positive-Well | Negative-Well | Concentration | Positive-Well | Negative-Well | Concentration | ||||||
| Jeju horse | J01 | 20,163 | 18,781 | 653 | 18,128 | 308.62 | 0 | 18,781 | 0 | 0 | Jeju horse |
| J02 | 20,163 | 18,430 | 615 | 17,815 | 295.98 | 0 | 18,430 | 0 | 0 | Jeju horse | |
| J03 | 20,163 | 17,464 | 624 | 16,840 | 317.31 | 0 | 17,464 | 0 | 0 | Jeju horse | |
| J04 | 20,163 | 18,389 | 615 | 17,774 | 296.65 | 0 | 18,389 | 0 | 0 | Jeju horse | |
| J05 | 20,163 | 17,790 | 258 | 17,532 | 127.4 | 255 | 17,535 | 125.91 | 1.01 | Heterozygote | |
| J06 | 20,163 | 16,825 | 414 | 16,411 | 217.27 | 0 | 16,825 | 0 | 0 | Jeju horse | |
| J07 | 20,163 | 18,703 | 256 | 18,447 | 120.19 | 264 | 18,439 | 123.98 | 0.97 | Heterozygote | |
| J08 | 20,163 | 17,508 | 539 | 16,969 | 272.7 | 0 | 17,508 | 0 | 0 | Jeju horse | |
| J09 | 20,163 | 18,835 | 549 | 18,286 | 257.97 | 0 | 18,835 | 0 | 0 | Jeju horse | |
| J10 | 20,163 | 19,167 | 733 | 18,434 | 340.06 | 0 | 19,167 | 0 | 0 | Jeju horse | |
| Thoroughbred | R01 | 20,163 | 18,413 | 0 | 18,413 | 0 | 213 | 18,200 | 101.47 | 0 | Thoroughbred |
| R02 | 20,163 | 17,219 | 0 | 17,219 | 0 | 154 | 17,065 | 78.35 | 0 | Thoroughbred | |
| R03 | 20,163 | 18,356 | 0 | 18,356 | 0 | 588 | 17,768 | 283.93 | 0 | Thoroughbred | |
| R04 | 20,163 | 18,365 | 228 | 18,137 | 108.95 | 256 | 18,109 | 122.42 | 0.89 | Heterozygote | |
| R05 | 20,163 | 18,890 | 0 | 18,890 | 0 | 655 | 18,235 | 307.76 | 0 | Thoroughbred | |
| R06 | 20,163 | 17,654 | 0 | 17,654 | 0 | 468 | 17,186 | 234.31 | 0 | Thoroughbred | |
| R07 | 20,163 | 19,244 | 0 | 19,244 | 0 | 535 | 18,709 | 245.88 | 0 | Thoroughbred | |
| R08 | 20,163 | 17,875 | 0 | 17,875 | 0 | 553 | 17,322 | 274.06 | 0 | Thoroughbred | |
| R09 | 20,163 | 18,125 | 0 | 18,125 | 0 | 486 | 17,639 | 237.03 | 0 | Thoroughbred | |
| R10 | 20,163 | 17,613 | 227 | 17,386 | 113.13 | 260 | 17,353 | 129.7 | 0.87 | Heterozygote | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, W.; Mun, S.; Choi, S.-Y.; Oh, D.-Y.; Park, Y.-S.; Han, K. Comparative Analysis for Genetic Characterization in Korean Native Jeju Horse. Animals 2021, 11, 1924. https://doi.org/10.3390/ani11071924
Lee W, Mun S, Choi S-Y, Oh D-Y, Park Y-S, Han K. Comparative Analysis for Genetic Characterization in Korean Native Jeju Horse. Animals. 2021; 11(7):1924. https://doi.org/10.3390/ani11071924
Chicago/Turabian StyleLee, Wooseok, Seyoung Mun, Song-Yi Choi, Dong-Yep Oh, Yong-Soo Park, and Kyudong Han. 2021. "Comparative Analysis for Genetic Characterization in Korean Native Jeju Horse" Animals 11, no. 7: 1924. https://doi.org/10.3390/ani11071924
APA StyleLee, W., Mun, S., Choi, S.-Y., Oh, D.-Y., Park, Y.-S., & Han, K. (2021). Comparative Analysis for Genetic Characterization in Korean Native Jeju Horse. Animals, 11(7), 1924. https://doi.org/10.3390/ani11071924