
Genome-Wide Identification of Candidate Genes for Milk Production Traits in Korean Holstein Cattle

 , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Phenotypes
2.2. Genotyping
2.3. Quality Control (QC) and Imputation
2.4. GWAS
2.4.1. Single Marker Regression (SMR)
2.4.2. Bayesian C (BayesC) Approach
2.5. Functional Annotations
3. Results
3.1. General Statistics
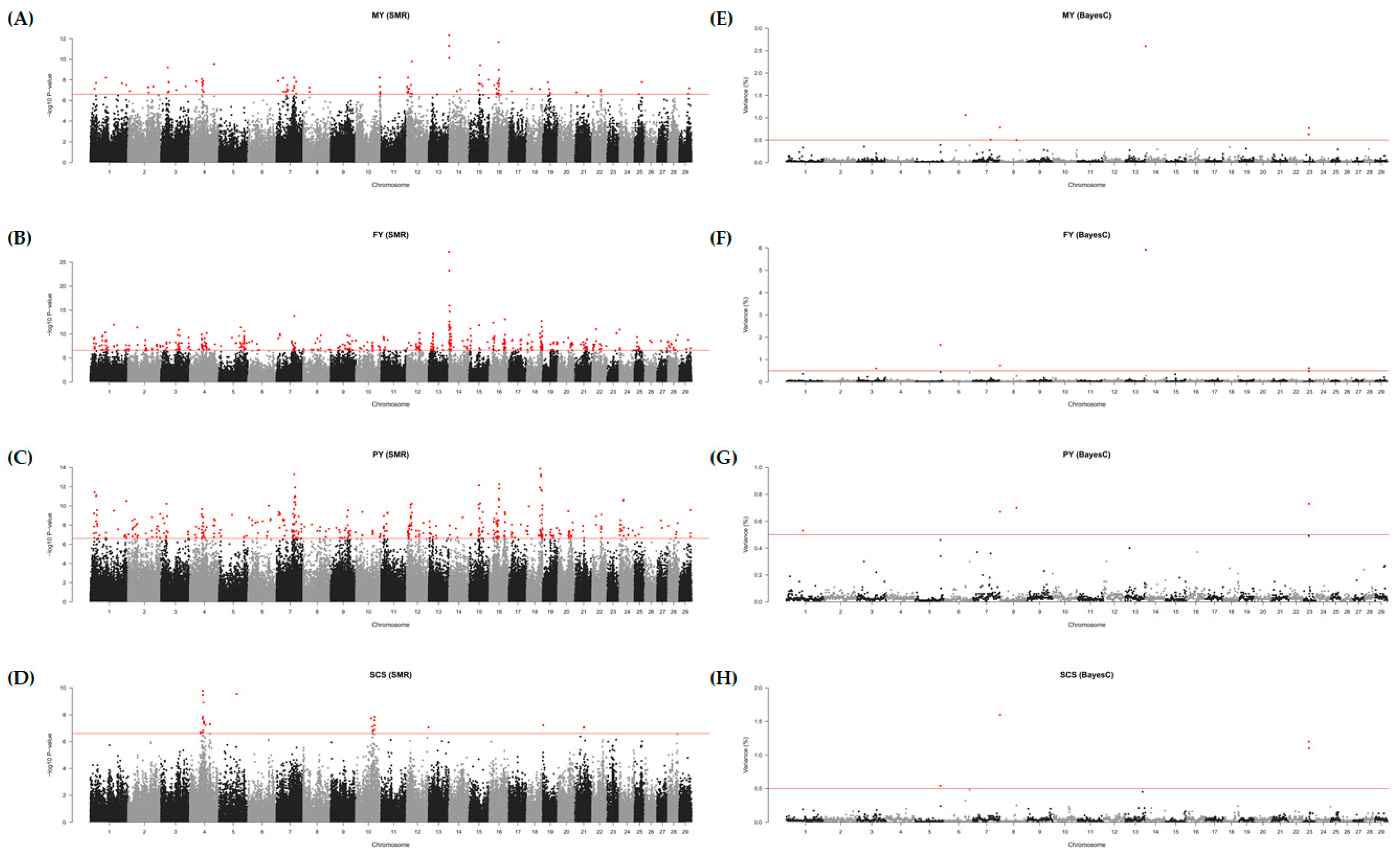
3.2. GWAS Based on the SMR
3.3. GWAS Based on the BayesC
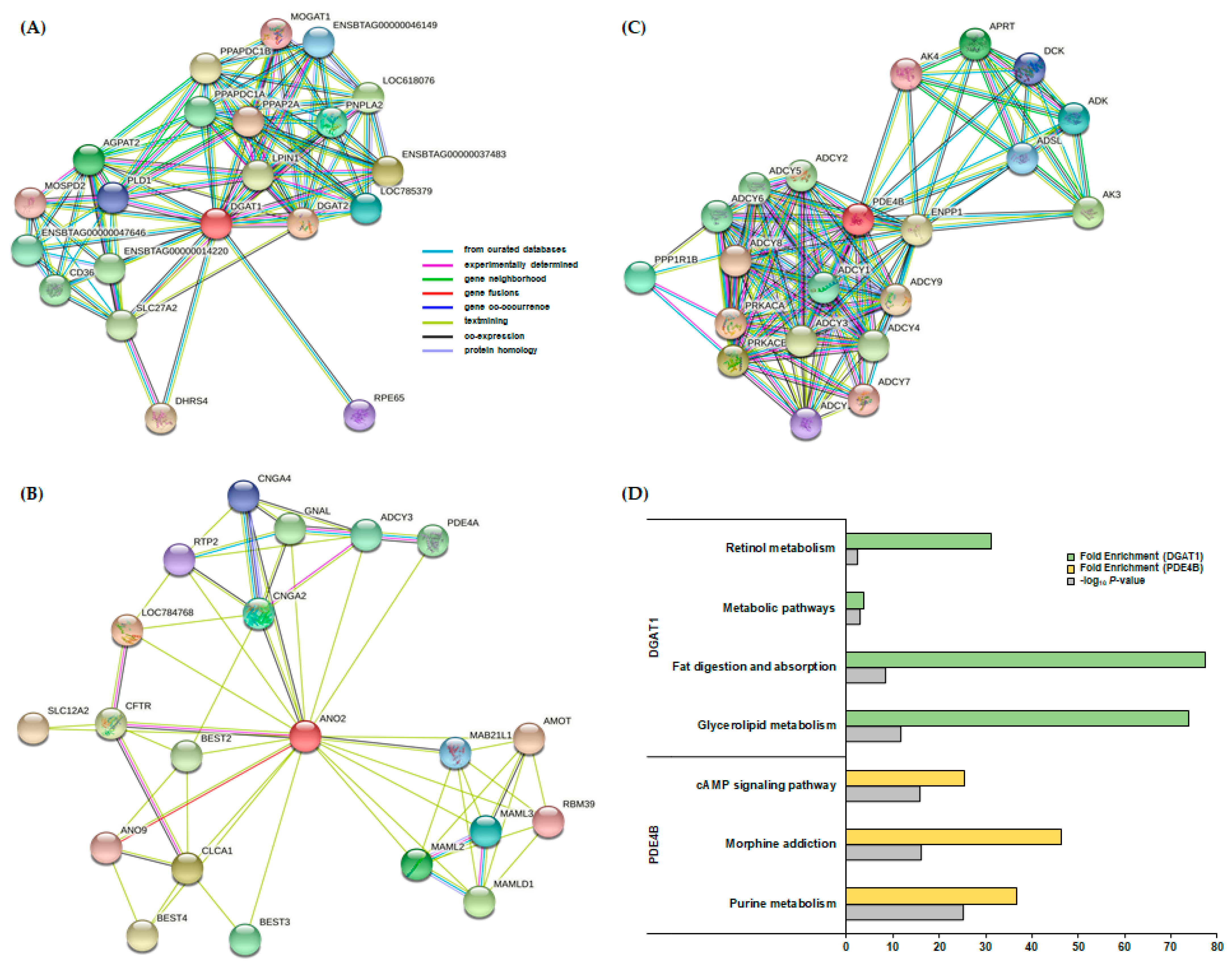
3.4. Fine Mapping and Candidate Genes
3.5. PPI and KEGG Pathway Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Henderson, C. Theoretical Basis and Computational Methods for a Number of Different Animal Models. J. Dairy Sci. 1988, 71, 1–16. [Google Scholar] [CrossRef]
- Georges, M.; Nielsen, D.; MacKinnon, M.; Mishra, A.; Okimoto, R.; Pasquino, A.T.; Sargeant, L.S.; Sorensen, A.; Steele, M.R.; Zhao, X. Mapping quantitative trait loci controlling milk production in dairy cattle by exploiting progeny testing. Genetics 1995, 139, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Sellner, E.; Kim, J.; McClure, M.; Taylor, K.; Schnabel, R.; Taylor, J. Board-invited review: Applications of genomic information in livestock. J. Anim. Sci. 2007, 85, 3148–3158. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.; Goddard, M.E. Prediction of identity by descent probabilities from marker-haplotypes. Genet. Sel. Evol. 2001, 33, 1–30. [Google Scholar] [CrossRef]
- Daw, E.W.; Heath, S.C.; Lu, Y. Single-nucleotide polymorphism versus microsatellite markers in a combined linkage and segregation analysis of a quantitative trait. BMC Genet. 2005, 6, S32–S33. [Google Scholar] [CrossRef][Green Version]
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef]
- Maxa, J.; Neuditschko, M.; Russ, I.; Förster, M.; Medugorac, I. Genome-wide association mapping of milk production traits in Braunvieh cattle. J. Dairy Sci. 2012, 95, 5357–5364. [Google Scholar] [CrossRef]
- Minozzi, G.; Nicolazzi, E.L.; Stella, A.; Biffani, S.; Negrini, R.; Lazzari, B.; Ajmone-Marsan, P.; Williams, J.L. Genome Wide Analysis of Fertility and Production Traits in Italian Holstein Cattle. PLoS ONE 2013, 8, e80219. [Google Scholar] [CrossRef]
- Nayeri, S.; Sargolzaei, M.; Abo-Ismail, M.K.; May, N.; Miller, S.P.; Schenkel, F.; Moore, S.S.; Stothard, P. Genome-wide association for milk production and female fertility traits in Canadian dairy Holstein cattle. BMC Genet. 2016, 17, 1–11. [Google Scholar] [CrossRef]
- Pausch, H.; Emmerling, R.; Gredler-Grandl, B.; Fries, R.; Daetwyler, H.D.; Goddard, M.E. Meta-analysis of sequence-based association studies across three cattle breeds reveals 25 QTL for fat and protein percentages in milk at nucleotide resolution. BMC Genom. 2017, 18, 853. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, J.; Chen, C.; Zhang, J.; Wen, W.; Tian, J.; Zhang, Z.; Gu, Y. GWAS-Based Identification of New Loci for Milk Yield, Fat, and Protein in Holstein Cattle. Animals 2020, 10, 2048. [Google Scholar] [CrossRef]
- Buitenhuis, B.; Janss, L.L.G.; Poulsen, N.A.; Larsen, L.B.; Larsen, M.K.; Sørensen, P. Genome-wide association and biological pathway analysis for milk-fat composition in Danish Holstein and Danish Jersey cattle. BMC Genom. 2014, 15, 1–11. [Google Scholar] [CrossRef]
- Strucken, E.M.; Bortfeldt, R.H.; Tetens, J.; Thaller, G.; Brockmann, G.A. Genetic effects and correlations between production and fertility traits and their dependency on the lactation-stage in Holstein Friesians. BMC Genet. 2012, 13, 108. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, J.; Sun, D.; Ma, P.; Ding, X.; Yu, Y.; Zhang, Q. Genome Wide Association Studies for Milk Production Traits in Chinese Holstein Population. PLoS ONE 2010, 5, e13661. [Google Scholar] [CrossRef]
- Wang, T.; Li, J.; Gao, X.; Song, W.; Chen, C.; Yao, D.; Ma, J.; Xu, L.; Ma, Y. Genome-wide association study of milk compo-nents in Chinese Holstein cows using single nucleotide polymorphism. Livest. Sci. 2020, 233, 103951. [Google Scholar] [CrossRef]
- Lee, S.; Dang, C.; Choy, Y.; Do, C.; Cho, K.; Kim, J.; Kim, Y.; Lee, J. Comparison of genome-wide association and genomic prediction methods for milk production traits in Korean Holstein cattle. Asian-Australas. J. Anim. Sci. 2019, 32, 913–921. [Google Scholar] [CrossRef]
- Shin, D.; Lee, C.; Park, K.-D.; Kim, H.; Cho, K.-H. Genome-association analysis of Korean Holstein milk traits using genomic estimated breeding value. Asian-Australas. J. Anim. Sci. 2015, 30, 309–319. [Google Scholar] [CrossRef]
- Li, Y.; Li, B.; Yang, M.; Han, H.; Chen, T.; Wei, Q.; Miao, Z.; Yin, L.; Wang, R.; Shen, J.; et al. Genome-Wide Association Study and Fine Mapping Reveals Candidate Genes for Birth Weight of Yorkshire and Landrace Pigs. Front. Genet. 2020, 11, 183. [Google Scholar] [CrossRef]
- Tribout, T.; Croiseau, P.; Lefebvre, R.; Barbat, A.; Boussaha, M.; Fritz, S.; Boichard, D.; Hoze, C.; Sanchez, M.-P. Confirmed effects of candidate variants for milk production, udder health, and udder morphology in dairy cattle. Genet. Sel. Evol. 2020, 52, 55. [Google Scholar] [CrossRef]
- Dutta, P.; Talenti, A.; Young, R.; Jayaraman, S.; Callaby, R.; Jadhav, S.K.; Dhanikachalam, V.; Manikandan, M.; Biswa, B.B.; Low, W.Y.; et al. Whole genome analysis of water buffalo and global cattle breeds highlights convergent signatures of domestication. Nature Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Fang, M.; Georges, M. BayesFM: A software program to fine-map multiple causative variants in GWAS identified risk loci. bioRxiv 2016, 067801. [Google Scholar] [CrossRef]
- Huang, H.; International Inflammatory Bowel Disease Genetics Consortium; Fang, M.; Jostins, L.; Mirkov, M.U.; Boucher, G.; Anderson, C.A.; Andersen, V.; Cleynen, I.; Cortes, A.; et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547, 173–178. [Google Scholar] [CrossRef]
- Kitano, H. Systems Biology: A Brief Overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef]
- Sanchez, M.-P.; Govignon-Gion, A.; Croiseau, P.; Fritz, S.; Hozé, C.; Miranda, G.; Martin, P.; Barbat-Leterrier, A.; Letaïef, R.; Rocha, D.; et al. Within-breed and multi-breed GWAS on imputed whole-genome sequence variants reveal candidate mutations affecting milk protein composition in dairy cattle. Genet. Sel. Evol. 2017, 49, 68. [Google Scholar] [CrossRef]
- Zhou, C.; Li, C.; Cai, W.; Liu, S.; Yin, H.; Shi, S.; Zhang, Q.; Zhang, S. Genome-wide association study for milk protein com-position traits in a Chinese Holstein population using a single-step approach. Front. Genet. 2019, 10, 72. [Google Scholar] [CrossRef]
- Du, C.; Deng, T.X.; Zhou, Y.; Ghanem, N.; Hua, G.H. Bioinformatics analysis of candidate genes for milk production traits in water buffalo (Bubalus bubalis). Trop. Anim. Health Prod. 2019, 52, 63–69. [Google Scholar] [CrossRef]
- Laodim, T.; Elzo, M.A.; Koonawootrittriron, S.; Suwanasopee, T.; Jattawa, D. Pathway enrichment and protein interaction network analysis for milk yield, fat yield and age at first calving in a Thai multibreed dairy population. Asian-Australas. J. Anim. Sci. 2018, 32, 508. [Google Scholar] [CrossRef]
- Palombo, V.; Milanesi, M.; Sgorlon, S.; Capomaccio, S.; Mele, M.; Nicolazzi, E.; Ajmone-Marsan, P.; Pilla, F.; Stefanon, B.; D’Andrea, M. Genome-wide association study of milk fatty acid composition in Italian Simmental and Italian Holstein cows using single nucleotide polymorphism arrays. J. Dairy Sci. 2018, 101, 11004–11019. [Google Scholar] [CrossRef]
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef]
- Saatchi, M.; Schnabel, R.D.; Rolf, M.M.; Taylor, J.F.; Garrick, D.J. Accuracy of direct genomic breeding values for nationally evaluated traits in US Limousin and Simmental beef cattle. Genet. Sel. Evol. 2012, 44, 38. [Google Scholar] [CrossRef]
- Sargolzaei, M.; Chesnais, J.P.; Schenkel, F.S. A new approach for efficient genotype imputation using information from rela-tives. BMC Genom. 2014, 15, 478. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Garrick, D.J.; Fernando, R.L. Implementing a QTL detection study (GWAS) using genomic prediction methodology. In Genome-Wide Association Studies and Genomic Prediction; Humana Press: Totowa, NJ, USA, 2013; pp. 275–298. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–Protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, 1–11. [Google Scholar] [CrossRef]
- Ponsuksili, S.; Reyer, H.; Trakooljul, N.; Murani, E.; Wimmers, K. Single-and Bayesian multi-marker genome-wide associa-tion for haematological parameters in pigs. PLoS ONE 2016, 11, e0159212. [Google Scholar] [CrossRef]
- Toosi, A.; Fernando, R.L.; Dekkers, J.C.M. Genome-wide mapping of quantitative trait loci in admixed populations using mixed linear model and Bayesian multiple regression analysis. Genet. Sel. Evol. 2018, 50, 32. [Google Scholar] [CrossRef]
- Yi, H.; Breheny, P.; Imam, N.; Liu, Y.; Hoeschele, I. Penalized multimarker vs. single-marker regression methods for ge-nome-wide association studies of quantitative traits. Genetics 2015, 199, 205–222. [Google Scholar] [CrossRef]
- Zare, Y.; Shook, G.E.; Collins, M.T.; Kirkpatrick, B.W. Genome-wide association analysis and genomic prediction of Myco-bacterium avium subspecies paratuberculosis infection in US Jersey cattle. PLoS ONE 2014, 9, e88380. [Google Scholar] [CrossRef]
- Gao, Y.; Dong, L.; Xu, S.; Xiao, S.; Fang, M.; Wang, Z. Genome-wide association study using single marker analysis and Bayesian methods for the gonadosomatic index in the large yellow croaker. Aquaculture 2018, 486, 26–30. [Google Scholar] [CrossRef]
- Li, J.; Das, K.; Fu, G.; Li, R.; Wu, R. The Bayesian lasso for genome-wide association studies. Bioinformatics 2010, 27, 516–523. [Google Scholar] [CrossRef]
- Xu, S. Estimating Polygenic Effects Using Markers of the Entire Genome. Genetics 2003, 163, 789–801. [Google Scholar] [CrossRef]
- Yi, N.; George, V.; Allison, D.B. Stochastic Search Variable Selection for Identifying Multiple Quantitative Trait Loci. Genetics 2003, 164, 1129–1138. [Google Scholar] [CrossRef]
- Ball, R.D. Bayesian Methods for Quantitative Trait Loci Mapping Based on Model Selection: Approximate Analysis Using the Bayesian Information Criterion. Genetics 2001, 159, 1351–1364. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, L.; Prakapenka, D.; VanRaden, P.M.; Cole, J.B.; Da, Y. A large-scale genome-wide association study in US Holstein cattle. Front. Genet. 2019, 10, 412. [Google Scholar] [CrossRef]
- Wang, X.; Ma, P.; Liu, J.; Zhang, Q.; Zhang, Y.; Ding, X.; Jiang, L.; Wang, Y.; Zhang, Y.; Sun, D.; et al. Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. 2015, 16, 1–9. [Google Scholar] [CrossRef]
- Ashwell, M.; Heyen, D.; Sonstegard, T.; Van Tassell, C.; Da, Y.; VanRaden, P.; Ron, M.; Weller, J.; Lewin, H. Detection of Quantitative Trait Loci Affecting Milk Production, Health, and Reproductive Traits in Holstein Cattle. J. Dairy Sci. 2004, 87, 468–475. [Google Scholar] [CrossRef]
- Coppieters, W.; Riquet, J.; Arranz, J.-J.; Berzi, P.; Cambisano, N.; Grisart, B.; Karim, L.; Marcq, F.; Moreau, L.; Nezer, C.; et al. A QTL with major effect on milk yield and composition maps to bovine Chromosome 14. Mamm. Genome 1998, 9, 540–544. [Google Scholar] [CrossRef]
- Heyen, D.W.; Weller, J.I.; Ron, M.; Band, M.; Beever, J.E.; Feldmesser, E.; Da, Y.; Wiggans, G.R.; VanRaden, P.M.; Lewin, H.A. A genome scan for QTL influencing milk production and health traits in dairy cattle. Physiol. Genom. 1999, 1, 165–175. [Google Scholar] [CrossRef][Green Version]
- Grisart, B.; Farnir, F.; Karim, L.; Cambisano, N.; Kim, J.-J.; Kvasz, A.; Mni, M.; Simon, P.; Frere, J.-M.; Coppieters, W.; et al. Genetic and functional confirmation of the causality of the DGAT1 K232A quantitative trait nucleotide in affecting milk yield and composition. Proc. Natl. Acad. Sci. USA 2004, 101, 2398–2403. [Google Scholar] [CrossRef]
- Wang, X.; Wurmser, C.; Pausch, H.; Jung, S.; Reinhardt, F.; Tetens, J.; Thaller, G.; Fries, R. Identification and Dissection of Four Major QTL Affecting Milk Fat Content in the German Holstein-Friesian Population. PLoS ONE 2012, 7, e40711. [Google Scholar] [CrossRef]
- Raven, L.-A.; Cocks, B.G.; Hayes, B.J. Multibreed genome wide association can improve precision of mapping causative variants underlying milk production in dairy cattle. BMC Genom. 2014, 15, 62. [Google Scholar] [CrossRef]
- Cases, S.; Smith, S.J.; Zheng, Y.-W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J. Identification of a gene encoding an acyl CoA: Diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef]
- Cheng, D.; Meegalla, R.L.; He, B.; Cromley, D.A.; Billheimer, J.T.; Young, P.R. Human acyl-CoA: Diacylglycerol acyltransferase is a tetrameric protein. Biochem. J. 2001, 359, 707–714. [Google Scholar] [CrossRef]
- Smith, S.J.; Cases, S.; Jensen, D.R.; Chen, H.C.; Sande, E.; Tow, B.; Sanan, D.A.; Raber, J.; Eckel, R.H.; Farese, R.V. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat. Genet. 2000, 25, 87–90. [Google Scholar] [CrossRef]
- Winter, A.; Krämer, W.; Werner, F.A.; Kollers, S.; Kata, S.; Durstewitz, G.; Buitkamp, J.; Womack, J.E.; Thaller, G.; Fries, R. Association of a lysine-232/alanine polymorphism in a bovine gene encoding acyl-CoA: Diacylglycerol acyltransferase (DGAT1) with variation at a quantitative trait locus for milk fat content. Proc. Natl. Acad. Sci. USA 2002, 99, 9300–9305. [Google Scholar] [CrossRef]
- Yen, C.-L.E.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Shin, D.; Lee, W.; Taye, M.; Cho, K.; Park, K.-D.; Kim, H. The prediction of the expected current selection coefficient of single nucleotide polymorphism associated with Holstein milk yield, fat and protein contents. Asian-Australas. J. Anim. Sci. 2016, 29, 36. [Google Scholar] [CrossRef]
- Kolbehdari, D.; Wang, Z.; Grant, J.R.; Murdoch, B.; Prasad, A.; Xiu, Z.; Marques, E.; Stothard, P.; Moore, S.S. A whole genome scan to map QTL for milk production traits and somatic cell score in Canadian Holstein bulls. J. Anim. Breed. Genet. 2009, 126, 216–227. [Google Scholar] [CrossRef]
- Boichard, D.; Grohs, C.; Bourgeois, F.; Cerqueira, F.; Faugeras, R.; Neau, A.; Rupp, R.; Amigues, Y.; Boscher, M.Y.; Levéziel, H. Detection of genes influencing economic traits in three French dairy cattle breeds. Genet. Sel. Evol. 2003, 35, 77–101. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Stierlé, A.; Beretz, A.; Schoeffter, P.; Lebec, A.; Wermuth, C.-G.; Cazenave, J.-P.; Stoclet, J.-C. Tissue and substrate specificity of inhibition by alkoxy-aryl-lactams of platelet and arterial smooth muscle cyclic nucleotide phosphodiesterases relationship to pharmacological activity. Biochem. Biophys. Res. Commun. 1983, 113, 954–959. [Google Scholar] [CrossRef]
- Lugnier, C.; Schoeffter, P.; Le Bec, A.; Strouthou, E.; Stoclet, J. Selective inhibition of cyclic nucleotide phosphodiesterases of human, bovine and rat aorta. Biochem. Pharmacol. 1986, 35, 1743–1751. [Google Scholar] [CrossRef]
- Conti, M.; Richter, W.; Mehats, C.; Livera, G.; Park, J.-Y.; Jin, C. Cyclic AMP-specific PDE4 Phosphodiesterases as Critical Components of Cyclic AMP Signaling. J. Biol. Chem. 2003, 278, 5493–5496. [Google Scholar] [CrossRef]
- Dostaler-Touchette, V.; Bédard, F.; Guillemette, C.; Pothier, F.; Chouinard, P.; Richard, F. Cyclic adenosine monophosphate (cAMP)-specific phosphodiesterase is functional in bovine mammary gland. J. Dairy Sci. 2009, 92, 3757–3765. [Google Scholar] [CrossRef]
- Omar, B.; Zmuda-Trzebiatowska, E.; Manganiello, V.; Göransson, O.; Degerman, E. Regulation of AMP-activated protein kinase by cAMP in adipocytes: Roles for phosphodiesterases, protein kinase B, protein kinase A, Epac and lipolysis. Cell. Signal. 2009, 21, 760–766. [Google Scholar] [CrossRef]
- Xie, W.; Ye, Y.; Feng, Y.; Xu, T.; Huang, S.; Shen, J.; Leng, Y. Linderane Suppresses Hepatic Gluconeogenesis by Inhibiting the cAMP/PKA/CREB Pathway Through Indirect Activation of PDE 3 via ERK/STAT3. Front. Pharmacol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Cochran, S.D.; Cole, J.B.; Null, D.J.; Hansen, P.J. Discovery of single nucleotide polymorphisms in candidate genes associated with fertility and production traits in Holstein cattle. BMC Genet. 2013, 14, 49. [Google Scholar] [CrossRef]
- Meredith, B.K.; Kearney, F.J.; Finlay, E.K.; Bradley, D.G.; Fahey, A.G.; Berry, D.P.; Lynn, D.J. Genome-wide associations for milk production and somatic cell score in Holstein-Friesian cattle in Ireland. BMC Genet. 2012, 13, 21. [Google Scholar] [CrossRef]
- Hinrichs, D.; Stamer, E.; Junge, W.; Kalm, E. Genetic Analyses of Mastitis Data Using Animal Threshold Models and Genetic Correlation with Production Traits. J. Dairy Sci. 2005, 88, 2260–2268. [Google Scholar] [CrossRef]
- Heringstad, B.; Gianola, D.; Chang, Y.; Ødegård, J.; Klemetsdal, G. Genetic associations between clinical mastitis and somatic cell score in early first-lactation cows. J. Dairy Sci. 2006, 89, 2236–2244. [Google Scholar] [CrossRef]
- Bloemhof, S.; de Jong, G.; de Haas, Y. Genetic parameters for clinical mastitis in the first three lactations of Dutch Holstein cattle. Vet. Microbiol. 2009, 134, 165–171. [Google Scholar] [CrossRef][Green Version]
- De Haas, Y.; Ouweltjes, W.; Napel, J.T.; Windig, J.; De Jong, G. Alternative Somatic Cell Count Traits as Mastitis Indicators for Genetic Selection. J. Dairy Sci. 2008, 91, 2501–2511. [Google Scholar] [CrossRef]
- Koivula, M.; Mäntysaari, E.; Negussie, E.; Serenius, T. Genetic and Phenotypic Relationships Among Milk Yield and Somatic Cell Count Before and After Clinical Mastitis. J. Dairy Sci. 2005, 88, 827–833. [Google Scholar] [CrossRef]
- Sender, G.; Korwin-Kossakowska, A.; Pawlik, A.; Hameed, K.G.A.; Oprządek, J. Genetic Basis of Mastitis Resistance in Dairy Cattle–A Review/Podstawy Genetyczne Odporności Krów Mlecznych Na Zapalenie Wymienia–Artykuł Przeglądowy. Ann. Anim. Sci. 2013, 13, 663–673. [Google Scholar] [CrossRef]
- Meredith, B.; Lynn, D.; Berry, D.; Kearney, F.; Bradley, D.; Finlay, E.; Fahey, A. A genome-wide association study for somatic cell score using the Illumina high-density bovine beadchip identifies several novel QTL potentially related to mastitis susceptibility. Front. Genet. 2013, 4, 229. [Google Scholar] [CrossRef]
- Tal-Stein, R.; Fontanesi, L.; Dolezal, M.; Scotti, E.; Bagnato, A.; Russo, V.; Canavesi, F.; Friedmann, A.; Soller, M.; Lipkin, E. A genome scan for QTL affecting milk somatic cell count in Israeli and Italian Holstein cows by means selective DNA pooling with multiple marker mapping. J. Dairy Sci. 2010, 93, 4913–4927. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PHENOTYPE (DEBV) a | BTA (Mb) b | GV (%) c | Informative SNP | Rs Number | Position d | SNP Effect | Model Frequency | Region Annotation | Gene Annotation |
|---|---|---|---|---|---|---|---|---|---|
| MY | 14 (1) | 2.60 | AX-371638654 | rs211016627 | 1,807,655 | 23.9100 | 0.1845 | Intron | HSF1 |
| AX-311625833 | rs384957047 | 1,793,616 | −22.1900 | 0.1717 | Upstream gene | DGAT1 (dist = 1735) | |||
| AX-311625843 | rs211223469 | 1,799,476 | −20.4900 | 0.1617 | Intron | DGAT1 | |||
| AX-419656711 | rs211282745 | 1,805,963 | −17.5500 | 0.1415 | Downstream gene | HSF1 (dist = 118) | |||
| AX-311625845 | rs209876151 | 1,800,439 | −15.9000 | 0.1300 | Intron | DGAT1 | |||
| AX-371657011 | rs208640292 | 1,806,875 | 12.8000 | 0.1095 | Synonymous | HSF1 | |||
| AX-419792758 | rs207655744 | 1,806,340 | 12.3300 | 0.1057 | 3 prime UTR | HSF1 | |||
| 6 (88) | 1.03 | AX-185121607 | rs110775601 | 88,952,089 | 78.4400 | 0.6617 | Intergenic | NPFFR2 (dist = 100,121) | |
| AX-106735408 | rs110527224 | 88,592,295 | 17.9900 | 0.1741 | Intergenic | SLC4A4 (dist = 54,249) | |||
| 8 (0) | 0.78 | AX-419764649 | rs721532493 | 887,406 | −1.0640 | 0.0183 | Intron | PALLD | |
| 23 (24) | 0.77 | AX-419655926 | rs380223715 | 24,021,950 | −1.3030 | 0.0180 | Intron | PKHD1 | |
| 23 (23) | 0.63 | AX-419634159 | rs517703887 | 23,999,941 | −1.0560 | 0.0169 | Intron | PKHD1 | |
| 7 (73) | 0.51 | AX-169404932 | rs135477609 | 73,561,312 | 4.0460 | 0.0477 | Intergenic | ADRA1B (dist = 49,805) | |
| 8 (69) | 0.50 | AX-419751453 | rs524049037 | 69,514,127 | −1.7210 | 0.0251 | Intron | GFRA2 | |
| FY | 14 (1) | 5.92 | AX-429953677 | rs110812136 | 1,991,225 | 1.9290 | 0.3670 | Intron | SPATC1 |
| AX-115099034 | rs109421300 | 1,801,116 | 1.5190 | 0.2793 | Intron | DGAT1 | |||
| AX-371657011 | rs208640292 | 1,806,875 | −1.1010 | 0.2097 | Synonymous | HSF1 | |||
| AX-419793247 | rs208317364 | 1,800,399 | −0.9766 | 0.1922 | Intron | DGAT1 | |||
| AX-419656711 | rs211282745 | 1,805,963 | 0.8508 | 0.1682 | Downstream gene | HSF1 (dist = 118) | |||
| AX-212342341 | rs135258919 | 1,808,145 | 0.8499 | 0.1693 | Missense | HSF1 | |||
| AX-419792758 | rs207655744 | 1,806,340 | −0.8430 | 0.1675 | 3 prime UTR | HSF1 | |||
| AX-117081655 | rs109234250 | 1,802,265 | −0.7752 | 0.1565 | Missense | DGAT1 | |||
| AX-124353826 | rs109326954 | 1,802,266 | −0.6848 | 0.1395 | Missense | DGAT1 | |||
| AX-311625843 | rs211223469 | 1,799,476 | 0.6695 | 0.1367 | Intron | DGAT1 | |||
| AX-311625845 | rs209876151 | 1,800,439 | 0.6313 | 0.1319 | Intron | DGAT1 | |||
| AX-311625833 | rs384957047 | 1,793,616 | 0.5941 | 0.1243 | Upstream gene | DGAT1 (dist = 1735) | |||
| AX-371638654 | rs211016627 | 1,807,655 | −0.4948 | 0.1052 | Intron | HSF1 | |||
| 5 (104) | 1.66 | AX-419663582 | rs43454033 | 104,831,727 | −0.1767 | 0.0560 | Intron | ANO2 | |
| 8 (0) | 0.74 | AX-419764649 | rs721532493 | 887,406 | −0.0319 | 0.0143 | Intron | PALLD | |
| 23 (24) | 0.62 | AX-419669189 | rs435871639 | 24,210,330 | −0.0168 | 0.0086 | Intron | PKHD1 | |
| 3 (79) | 0.60 | AX-106724308 | rs42314807 | 79,480,234 | −1.8050 | 0.4151 | Intron | PDE4B | |
| AX-169413290 | rs41596885 | 79,508,402 | 0.7012 | 0.1766 | Intron | PDE4B | |||
| PY | 23 (24) | 0.73 | AX-419655926 | rs380223715 | 24,021,950 | −0.0118 | 0.0086 | Intron | PKHD1 |
| 8 (69) | 0.70 | AX-419606850 | rs211419403 | 69,542,993 | −0.0259 | 0.0144 | Intron | GFRA2 | |
| 8 (0) | 0.67 | AX-419764649 | rs721532493 | 887,406 | −0.0214 | 0.0137 | Intron | PALLD | |
| 1 (69) | 0.53 | AX-419771850 | rs799074643 | 69,736,662 | 0.0607 | 0.0294 | Intron | UMPS | |
| SCS | 8 (0) | 1.60 | AX-419631051 | rs109008410 | 668,048 | 0.0012 | 0.0253 | Intron | PALLD |
| 23 (23) | 1.20 | AX-312701115 | rs467721520 | 23,807,184 | −0.0006 | 0.0150 | Intron | PKHD1 | |
| 23 (24) | 1.10 | AX-106721976 | rs109825181 | 24,117,682 | −0.0012 | 0.0246 | Intron | PKHD1 | |
| 5 (104) | 0.54 | AX-124344695 | rs110571898 | 104,682,238 | 0.0005 | 0.0127 | Missense | VWF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Lim, B.; Cho, J.; Lee, S.; Dang, C.-G.; Jeon, J.-H.; Kim, J.-M.; Lee, J. Genome-Wide Identification of Candidate Genes for Milk Production Traits in Korean Holstein Cattle. Animals 2021, 11, 1392. https://doi.org/10.3390/ani11051392
Kim S, Lim B, Cho J, Lee S, Dang C-G, Jeon J-H, Kim J-M, Lee J. Genome-Wide Identification of Candidate Genes for Milk Production Traits in Korean Holstein Cattle. Animals. 2021; 11(5):1392. https://doi.org/10.3390/ani11051392
Chicago/Turabian StyleKim, Sangwook, Byeonghwi Lim, Joohyeon Cho, Seokhyun Lee, Chang-Gwon Dang, Jung-Hwan Jeon, Jun-Mo Kim, and Jungjae Lee. 2021. "Genome-Wide Identification of Candidate Genes for Milk Production Traits in Korean Holstein Cattle" Animals 11, no. 5: 1392. https://doi.org/10.3390/ani11051392
APA StyleKim, S., Lim, B., Cho, J., Lee, S., Dang, C.-G., Jeon, J.-H., Kim, J.-M., & Lee, J. (2021). Genome-Wide Identification of Candidate Genes for Milk Production Traits in Korean Holstein Cattle. Animals, 11(5), 1392. https://doi.org/10.3390/ani11051392