
Inbreeding Calculated with Runs of Homozygosity Suggests Chromosome-Specific Inbreeding Depression Regions in Line 1 Hereford

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaeffer, L.R. Strategy for applying genome-wide selection in dairy cattle. J. Anim. Breed. Genet. 2006, 123, 218–223. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.; Van Tassell, C.; Wiggans, G.; Sonstegard, T.S.; Schnabel, R.; Taylor, J.; Schenkel, F. Invited Review: Reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 2009, 92, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Harris, B.; Johnson, D. Genomic predictions for New Zealand dairy bulls and integration with national genetic evaluation. J. Dairy Sci. 2010, 93, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Su, G.; Christensen, O.F.; Ostersen, T.; Henryon, M.; Lund, M.S. Estimating Additive and Non-Additive Genetic Variances and Predicting Genetic Merits Using Genome-Wide Dense Single Nucleotide Polymorphism Markers. PLoS ONE 2012, 7, e45293. [Google Scholar] [CrossRef]
- MacNeil, M.D. Invited Review: Research contributions from seventy-five years of breeding Line 1 Hereford cattle at Miles City, Montana1,2. J. Anim. Sci. 2009, 87, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, M.D.; Urick, J.J.; Newman, S.; Knapp, B.W. Selection for postweaning growth in inbred Hereford cattle: The Fort Keogh, Montana line 1 example. J. Anim. Sci. 1992, 70, 723–733. [Google Scholar] [CrossRef]
- Sumreddee, P.; Toghiani, S.; Hay, E.H.; Roberts, A.; E Agrrey, S.; Rekaya, R.; Togniani, S.; E Aggrey, S. Inbreeding depression in line 1 Hereford cattle population using pedigree and genomic information1. J. Anim. Sci. 2018, 97, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lozada-Soto, E.A.; Maltecca, C.; Lu, D.; Miller, S.; Cole, J.B.; Tiezzi, F. Trends in genetic diversity and the ef-fect of inbreeding in American Angus cattle under genomic selection. Genet. Sel. Evol. 2021, 53, 1–15. [Google Scholar] [CrossRef]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of au-tozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalls, M.A.; Guerreiro, R.J.; Simon-Sanchez, J.; Bras, J.T.; Traynor, B.J.; Gibbs, J.R.; Launer, L.; Hardy, J.; Singleton, A.B. Extended tracts of homozygosity identify novel candidate genes associated with late-onset Alzheimer’s dis-ease. Neurogenetics 2009, 10, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.T.; Tiezzi, F.; Huang, Y.; Gray, K.A.; Maltecca, C. A heuristic method to identify runs of homozy-gosity associated with reduced performance in livestock. J. Anim. Sci. 2017, 95, 4318–4332. [Google Scholar] [CrossRef] [Green Version]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Sargolzaei, M.; Chesnais, J.P.; Schenkel, F.S. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Maltecca, C.; Cassady, J.P.; Alexander, L.J.; Snelling, W.M.; MacNeil, M.D. Effects of reduced panel, reference origin, and genetic relationship on imputation of genotypes in Hereford cattle. J. Anim. Sci. 2012, 90, 4203–4208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Maltecca, C.; MacNeil, M.D.; Alexander, L.J.; Snelling, W.; Cassady, J.P. Using 50 k single nucleo-tide polymorphisms to elucidate genomic architecture of line 1 Hereford cattle. Front. Genet. 2012, 3, 285. [Google Scholar] [CrossRef] [Green Version]
- Coster, A. Pedigree: Pedigree Functions. R Package Version 1.4. 2012. Available online: https://CRAN.R-project.org/package=pedigree (accessed on 24 October 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 24 October 2021).
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. R Package Version 0.9.6. 2019. Available online: https://CRAN.R-project.org/package=detectRUNS (accessed on 24 October 2021).
- Tukey, J.W. Comparing Individual Means in the Analysis of Variance. Biometrics 1949, 5, 99. [Google Scholar] [CrossRef]
- Pryce, J.E.; Haile-Mariam, M.; Goddard, M.E.; Hayes, B.J. Identification of genomic regions associated with inbreeding depression in Holstein and Jersey dairy cattle. Genet. Sel. Evol. 2014, 46, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, J.T.; Haile-Mariam, M.; Pryce, J.E.; Maltecca, C. Investigation of regions impacting inbreeding de-pression and their association with the additive genetic effect for United States and Australia Jersey dairy cattle. BMC Genom. 2015, 16, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.L.; Fritz, E.R.; Reecy, J.M. AnimalQTLdb: A livestock QTL database tool set for positional QTL infor-mation mining and beyond. Nucleic Acids Res. 2007, 35 (Suppl. 1), D604–D609. [Google Scholar] [CrossRef]
- Baes, C.F.; Makanjuola, B.O.; Miglior, F.; Marras, G.; Howard, J.T.; Fleming, A.; Maltecca, C. Symposium re-view: The genomic architecture of inbreeding: How homozygosity affects health and performance. J. Dairy Sci. 2019, 102, 2807–2817. [Google Scholar] [CrossRef]
- Saatchi, M.; E Beever, J.; E Decker, J.; Faulkner, D.B.; Freetly, H.C.; Hansen, S.L.; Yampara-Iquise, H.; A Johnson, K.; Kachman, S.D.; Kerley, M.S.; et al. QTLs associated with dry matter intake, metabolic mid-test weight, growth and feed efficiency have little overlap across 4 beef cattle studies. BMC Genom. 2014, 15, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Akanno, E.C.; Plastow, G.; Fitzsimmons, C.; Miller, S.P.; Baron, V.; Ominski, K.; Basarab, J.A. Genome-wide association for heifer reproduction and calf performance traits in beef cattle. Genome 2015, 58, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.G.; Fitzsimons, C.; McClure, M.C.; McKenna, C.; Conroy, S.; Kenny, D.A.; McGee, M.; Waters, S.M.; Morris, D.W. GWAS and eQTL analysis identifies a SNP associated with both residual feed intake and GFRA2 ex-pression in beef cattle. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seabury, C.M.; Oldeschulte, D.L.; Saatchi, M.; Beever, J.E.; Decker, J.E.; Halley, Y.A.; Bhattarai, E.K.; Molaei, M.; Freetly, H.C.; Hansen, S.L.; et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genom. 2017, 18, 1–25. [Google Scholar] [CrossRef] [Green Version]
- An, B.; Xu, L.; Xia, J.; Wang, X.; Miao, J.; Chang, T.; Song, M.; Ni, J.; Xu, L.; Zhang, L.; et al. Multiple associa-tion analysis of loci and candidate genes that regulate body size at three growth stages in Simmental beef cattle. BMC Genet. 2020, 21, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltecca, C.; Tiezzi, F.; Cole, J.B.; Baes, C. Symposium review: Exploiting homozygosity in the era of genomics—Selection, inbreeding, and mating programs. J. Dairy Sci. 2020, 103, 5302–5313. [Google Scholar]
- Hedrick, P.W. Purging inbreeding depression and the probability of extinction: Full-sib mating. Heredity 1994, 73, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Macciotta, N.P.; Colli, L.; Cesarani, A.; Ajmone-Marsan, P.; Low, W.Y.; Tearle, R.; Williams, J.L. The distribu-tion of runs of homozygosity in the genome of river and swamp buffaloes reveals a history of adaptation, migration and crossbred events. Genet. Sel. Evol. 2021, 53, 1–21. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Reverter, A.; Porto-Neto, L.R.; Fortes, M.R.S.; Kasarapu, P.; De Cara, M.A.R.; Burrow, H.M.; Lehnert, S.A. Genomic inbreeding depression for climatic adaptation of tropical beef cattle. J. Anim. Sci. 2017, 95, 3809–3821. [Google Scholar] [CrossRef]
- Hidalgo, J.; Cesarani, A.; Garcia, A.; Sumreddee, P.; Larios, N.; Mancin, E.; García, J.; Núñez, R.; Ramírez, R. Genetic Background and Inbreeding Depression in Romosinuano Cattle Breed in Mexico. Animals 2021, 11, 321. [Google Scholar] [CrossRef]
- Fragomeni, B.O.; Lourenco, D.A.L.; Masuda, Y.; Legarra, A.; Misztal, I. Incorporation of causative quantitative trait nucleotides in single-step GBLUP. Genet. Sel. Evol. 2017, 49, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fragomeni, B.O.; Lourenco, D.A.L.; Legarra, A.; VanRaden, P.M.; Misztal, I. Alternative SNP weighting for single-step genomic best linear unbiased predictor evaluation of stature in US Holsteins in the presence of selected se-quence variants. J. Dairy Sci. 2019, 102, 10012–10019. [Google Scholar] [CrossRef] [PubMed]
- Brøndum, R.F.; Su, G.; Janss, L.; Sahana, G.; Guldbrandtsen, B.; Boichard, D.; Lund, M.S. Quantitative trait loci markers derived from whole genome sequence data increases the reliability of genomic prediction. J. Dairy Sci. 2015, 98, 4107–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, I.M.; Bowman, P.J.; Jagt, C.J.V.; Haile-Mariam, M.; Kemper, K.E.; Chamberlain, A.J.; Schrooten, C.; Hayes, B.J.; Goddard, M.E. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doekes, H.P.; Bijma, P.; Veerkamp, R.F.; De Jong, G.; Wientjes, Y.C.J.; Windig, J.J. Inbreeding depression across the genome of Dutch Holstein Friesian dairy cattle. Genet. Sel. Evol. 2020, 52, 1–18. [Google Scholar] [CrossRef]
- Cesarani, A.; Gaspa, G.; Pauciullo, A.; Degano, L.; Vicario, D.; Macciotta, N. P Genome-wide analysis of ho-mozygosity regions in european simmental bulls. J. Anim. Breed. Genet. 2021, 138, 69–79. [Google Scholar] [CrossRef]
- Martikainen, K.; Sironen, A.; Uimari, P. Estimation of intrachromosomal inbreeding depression on female fer-tility using runs of homozygosity in Finnish Ayrshire cattle. J. Dairy Sci. 2018, 101, 11097–11107. [Google Scholar] [CrossRef] [Green Version]
- Browning, S.R.; Browning, B.L. Identity by Descent Between Distant Relatives: Detection and Applications. Annu. Rev. Genet. 2012, 46, 617–633. [Google Scholar] [CrossRef]
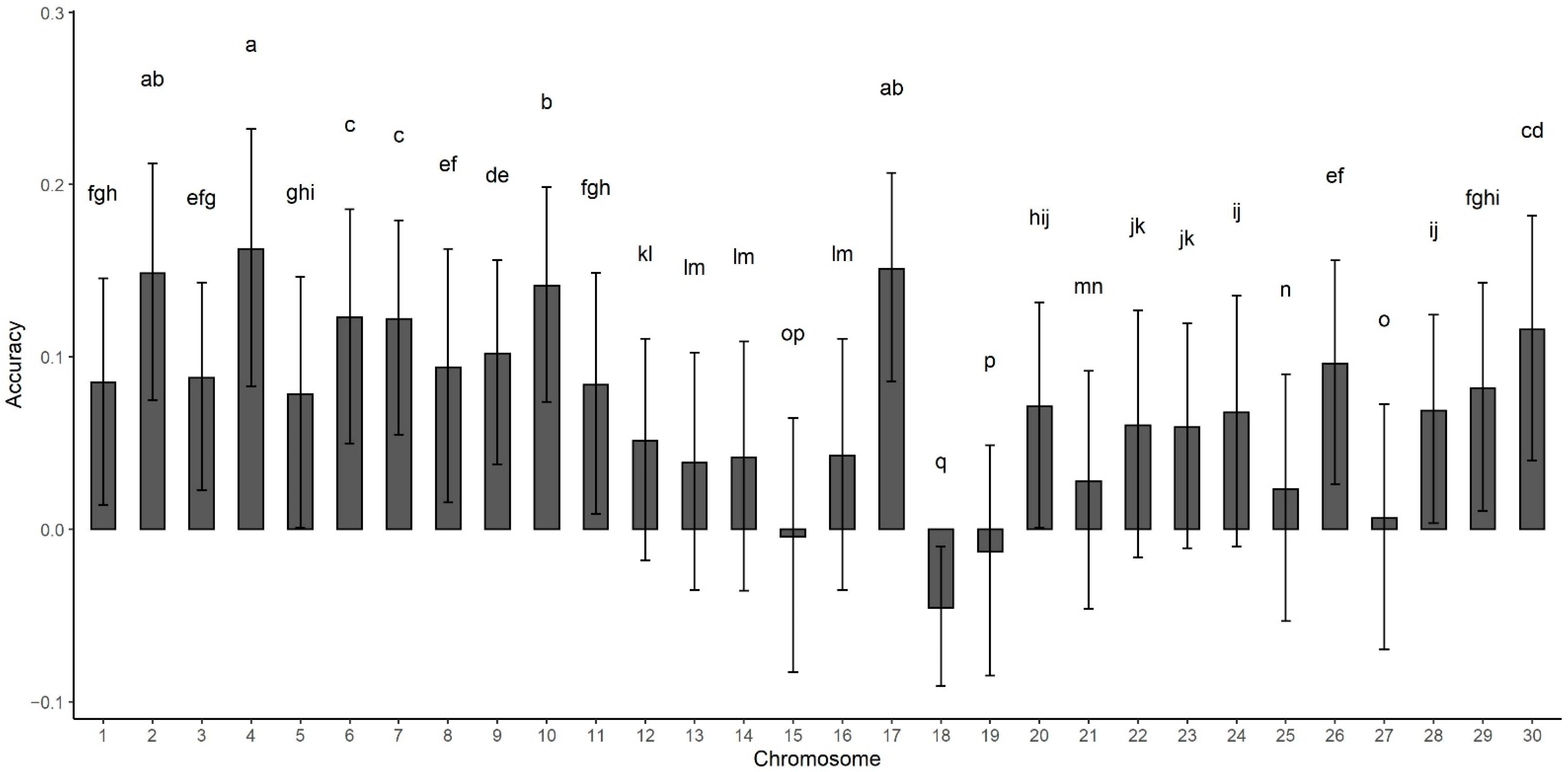
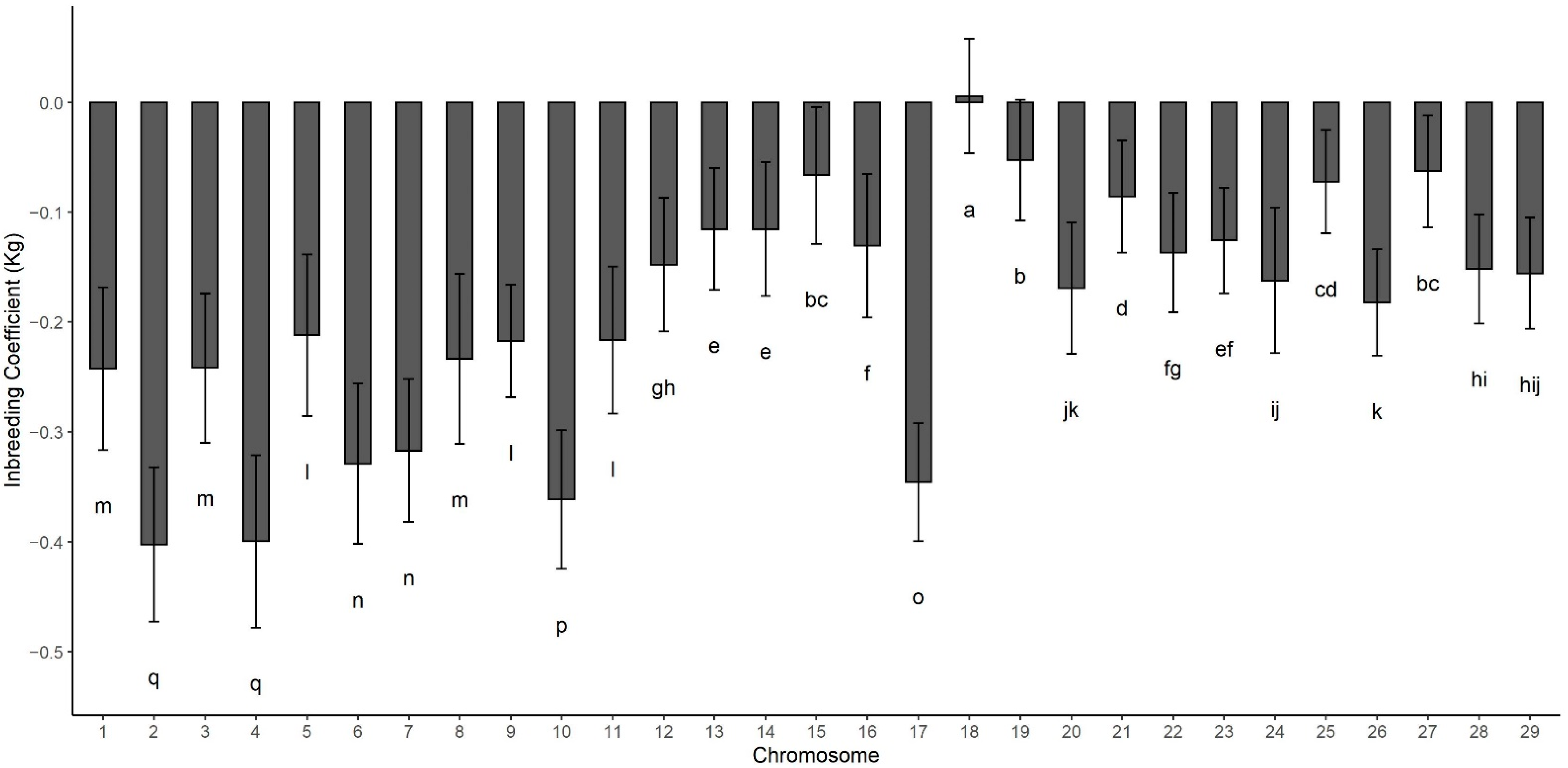

{kind=link}
{kind=link}
| Model * | Accuracy | SD |
| ADG = FPED | 0.17 e | 0.05 |
| ADG = FPED + Sex | 0.76 c | 0.02 |
| ADG = FPED + Sex + YOB | 0.88 a | 0.02 |
| ADG = FROH | 0.21 d | 0.05 |
| ADG = FROH + Sex | 0.77 b | 0.02 |
| ADG = FROH + Sex + YOB | 0.88 a | 0.02 |
| ADG = Sex | 0.76 c | 0.03 |
| ADG = Sex + YOB | 0.88 a | 0.02 |
| ADGC = FPED | 0.01 f | 0.3 |
| ADGC = FROH | 0.02 g | 0.4 |
| Model | Accuracy | SD |
|---|---|---|
| ADGC~CHR1-CHRX | 0.13 | 0.06 |
| ADGC~Chr1 + Chr4 + Chr6 + Chr7 + Chr10 + Chr15 + Chr17 + Chr22 | 0.19 | 0.06 |
| ADGC~Chr1 + Chr4 + Chr6 + Chr7 + Chr10 + Chr15 + Chr17 + Chr22 + ChrX | 0.19 | 0.06 |
| ADGC~Chr4 + Chr7 + Chr10 + Chr15 + Chr17 + Chr18 + ChrX | 0.22 | 0.06 |
| ADGC~Chr7 + Chr10 + Chr17 + Chr18 + ChrX | 0.21 | 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilon, B.; Hinterneder, K.; Hay, E.H.A.; Fragomeni, B. Inbreeding Calculated with Runs of Homozygosity Suggests Chromosome-Specific Inbreeding Depression Regions in Line 1 Hereford. Animals 2021, 11, 3105. https://doi.org/10.3390/ani11113105
Pilon B, Hinterneder K, Hay EHA, Fragomeni B. Inbreeding Calculated with Runs of Homozygosity Suggests Chromosome-Specific Inbreeding Depression Regions in Line 1 Hereford. Animals. 2021; 11(11):3105. https://doi.org/10.3390/ani11113105
Chicago/Turabian StylePilon, Bethany, Kelly Hinterneder, El Hamidi A. Hay, and Breno Fragomeni. 2021. "Inbreeding Calculated with Runs of Homozygosity Suggests Chromosome-Specific Inbreeding Depression Regions in Line 1 Hereford" Animals 11, no. 11: 3105. https://doi.org/10.3390/ani11113105
APA StylePilon, B., Hinterneder, K., Hay, E. H. A., & Fragomeni, B. (2021). Inbreeding Calculated with Runs of Homozygosity Suggests Chromosome-Specific Inbreeding Depression Regions in Line 1 Hereford. Animals, 11(11), 3105. https://doi.org/10.3390/ani11113105