On the Variability of Microbial Populations and Bacterial Metabolites within the Canine Stool. An in-Depth Analysis



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. Stool Subsampling
2.3. Stool Preparation for Chemical Analysis and pH Determination
2.4. Chemical Analysis
2.5. Microbial Analysis
2.6. Statistical Analysis
3. Results
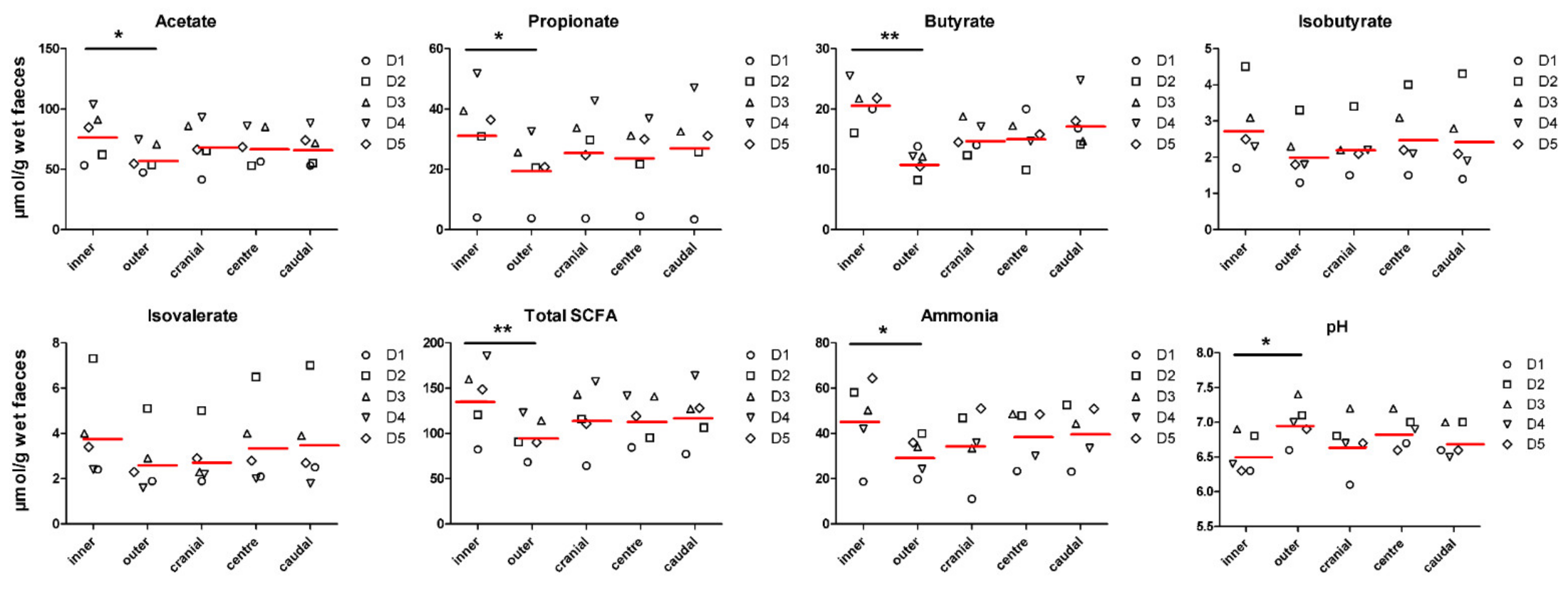
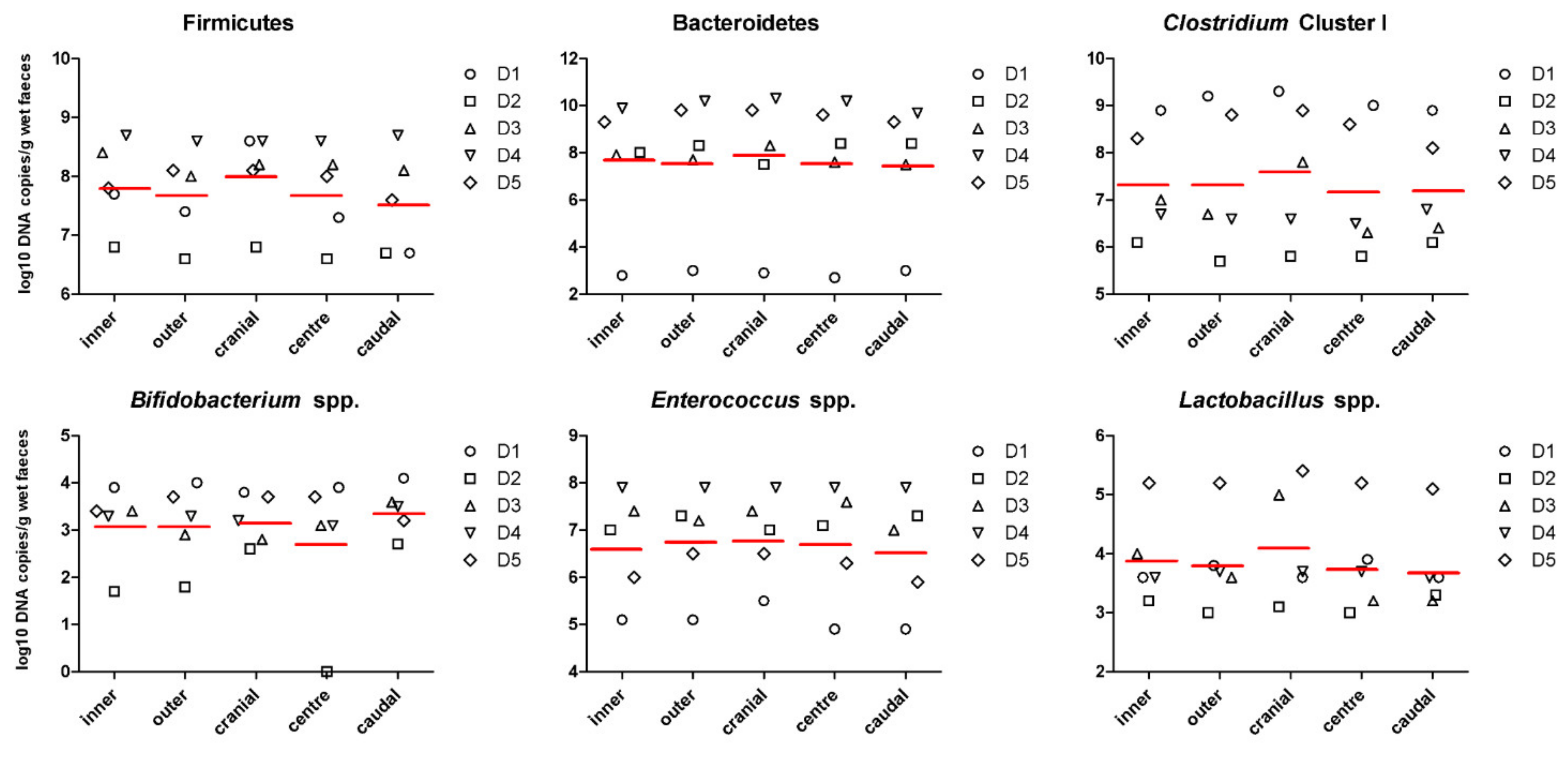
3.1. Faecal Concentration of Bacterial Metabolites and pH Are Affected by Inner and Outer Subsampling but Not by Longitudinal Subsampling
3.2. Homogenisation Has Minimal Effect on Metabolites Concentration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Data Availability Statement
Conflicts of Interest
References
- Pilla, R.; Suchodolski, J.S. The role of the canine gut microbiome and metabolome in health and gastrointestinal disease. Front. Vet. Sci. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Swanson, K.S. Gut microbiota of humans, dogs and cats: Current knowledge and future opportunities and challenges. Br. J. Nutr. 2015, 113, S6–S17. [Google Scholar] [CrossRef] [PubMed]
- Suchodolski, J.S. Diagnosis and interpretation of intestinal dysbiosis in dogs and cats. Vet. J. 2016, 215, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Suchodolski, J.S.; Dowd, S.E.; Wilke, V.; Steiner, J.M.; Jergens, A.E. 16S rRNA gene pyrosequencing reveals bacterial dysbiosis in the duodenum of dogs with idiopathic inflammatory bowel disease. PLoS ONE 2012, 7, e39333. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, F.; Minamoto, Y.; Suchodolski, J.S.; Galiazzo, G.; Vecchiato, C.G.; Pinna, C.; Biagi, G.; Pietra, M. Effect of an extruded animal protein-free diet on fecal microbiota of dogs with food-responsive enteropathy. J. Vet. Intern. Med. 2018, 32, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Guard, B.C.; Barr, J.W.; Reddivari, L.; Klemashevich, C.; Jayaraman, A.; Steiner, J.M.; Vanamala, J.; Suchodolski, J.S. Characterization of microbial dysbiosis and metabolomic changes in dogs with acute diarrhea. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Unterer, S.; Busch, K.; Leipig, M.; Hermanns, W.; Wolf, G.; Straubinger, R.K.; Mueller, R.S.; Hartmann, K. Endoscopically visualized lesions, histologic findings, and bacterial invasion in the gastrointestinal mucosa of dogs with acute hemorrhagic diarrhea syndrome. J. Vet. Intern. Med. 2014, 28, 52–58. [Google Scholar] [CrossRef]
- Garcia-Mazcorro, J.F.; Barcenas-Walls, J.R.; Suchodolski, J.S.; Steiner, J.M. Molecular assessment of the fecal microbiota in healthy cats and dogs before and during supplementation with fructo-oligosaccharides (FOS) and inulin using high-throughput 454- pyrosequencing. PeerJ 2017, 2017, e3184. [Google Scholar] [CrossRef]
- Handl, S.; German, A.J.; Holden, S.L.; Dowd, S.E.; Steiner, J.M.; Heilmann, R.M.; Grant, R.W.; Swanson, K.S.; Suchodolski, J.S. Faecal microbiota in lean and obese dogs. FEMS Microbiol. Ecol. 2013, 84, 332–343. [Google Scholar] [CrossRef]
- Ezzy, A.C.; Hagstrom, A.D.; George, C.; Hamlin, A.S.; Pereg, L.; Murphy, A.J.; Winter, G. Storage and handling of human faecal samples affect the gut microbiome composition: A feasibility study. J. Microbiol. Methods 2019, 164, 105668. [Google Scholar] [CrossRef]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.-E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Van Zyl, K.N.; Whitelaw, A.C.; Newton-Foot, M. The effect of storage conditions on microbial communities in stool. PLoS ONE 2020, 15, e0227486. [Google Scholar] [CrossRef] [PubMed]
- Conrads, G.; Abdelbary, M.M.H. Challenges of next-generation sequencing targeting anaerobes. Anaerobe 2019, 58, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Laudadio, I.; Fulci, V.; Stronati, L.; Carissimi, C. Next-Generation Metagenomics: Methodological challenges and opportunities. Omics J. Integr. Biol. 2019, 23, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Gorzelak, M.A.; Gill, S.K.; Tasnim, N.; Ahmadi-Vand, Z.; Jay, M.; Gibson, D.L. Methods for improving human gut microbiome data by reducing variability through sample processing and storage of stool. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Videnska, P.; Smerkova, K.; Zwinsova, B.; Popovici, V.; Micenkova, L.; Sedlar, K.; Budinska, E. Stool sampling and DNA isolation kits affect DNA quality and bacterial composition following 16S rRNA gene sequencing using MiSeq Illumina platform. Sci. Rep. 2019, 9, 13837. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Chen, Z.; Hui, M.; Wong, M.C.S.; Ho, W.C.S.; Chin, M.L.; Ng, S.C.; Chan, F.K.L.; Chan, P.K.S. Impact of inter- and intra-individual variation, sample storage and sampling fraction on human stool microbial community profiles. PeerJ 2019, 7, e6172. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Peterson, C.M.; Raggio, A.; Keenan, M.J.; Martin, R.J.; Ravussin, E.; Marco, M.L. Impact of different fecal processing methods on assessments of bacterial diversity in the human intestine. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Santiago, A.; Panda, S.; Mengels, G.; Martinez, X.; Azpiroz, F.; Dore, J.; Guarner, F.; Manichanh, C. Processing faecal samples: A step forward for standards in microbial community analysis. BMC Microbiol. 2014, 14, 112. [Google Scholar] [CrossRef]
- Lin, C.Y.; Cross, T.W.L.; Doukhanine, E.; Swanson, K.S. An ambient temperature collection and stabilization strategy for canine microbiota studies. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Gratton, J.; Phetcharaburanin, J.; Mullish, B.H.; Williams, H.R.T.; Thursz, M.; Nicholson, J.K.; Holmes, E.; Marchesi, J.R.; Li, J.V. Optimized sample handling strategy for metabolic profiling of human feces. Anal. Chem. 2016, 88, 4661–4668. [Google Scholar] [CrossRef] [PubMed]
- Pinna, C.; Vecchiato, C.G.; Bolduan, C.; Grandi, M.; Stefanelli, C.; Windisch, W.; Zaghini, G.; Biagi, G. Influence of dietary protein and fructooligosaccharides on fecal fermentative end-products, fecal bacterial populations and apparent total tract digestibility in dogs. BMC Vet. Res. 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Pinna, C.; Vecchiato, C.G.; Grandi, M.; Mammi, L.M.E.; Stefanelli, C.; Biagi, G. In vitro evaluation of the effects of tylosin on the composition and metabolism of canine fecal microbiota. Animals 2020, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Mühling, M.; Woolven-Allen, J.; Murrell, J.C.; Joint, I. Improved group-specific PCR primers for denaturing gradient gel electrophoresis analysis of the genetic diversity of complex microbial communities. ISME J. 2008, 2, 379–392. [Google Scholar] [CrossRef]
- Song, Y.; Liu, C.; Finegold, S.M. Real-time PCR quantitation of clostridia in feces of autistic children. Appl. Environ. Microbiol. 2004, 70, 6459–6465. [Google Scholar] [CrossRef]
- Malinen, E.; Rinttilä, T.; Kajander, K.; Mättö, J.; Kassinen, A.; Krogius, L.; Saarela, M.; Korpela, R.; Palva, A. Analysis of the fecal microbiota of irritable bowel syndrome patients and healthy controls with real-time PCR. Am. J. Gastroenterol. 2005, 100, 373–382. [Google Scholar] [CrossRef]
- Rinttilä, T.; Kassinen, A.; Malinen, E.; Krogius, L.; Palva, A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J. Appl. Microbiol. 2004, 97, 1166–1177. [Google Scholar] [CrossRef]
- Pereira, A.M.; Pinna, C.; Biagi, G.; Stefanelli, C.; Maia, M.R.G.; Matos, E.; Segundo, M.A.; Fonseca, A.J.M.; Cabrita, A.R.J. Supplemental selenium source on gut health: Insights on fecal microbiome and fermentation products of growing puppies. FEMS Microbiol. Ecol. 2020, 96. [Google Scholar] [CrossRef]
- Minamoto, Y.; Minamoto, T.; Isaiah, A.; Sattasathuchana, P.; Buono, A.; Rangachari, V.R.; McNeely, I.H.; Lidbury, J.; Steiner, J.M.; Suchodolski, J.S. Fecal short-chain fatty acid concentrations and dysbiosis in dogs with chronic enteropathy. J. Vet. Intern. Med. 2019, 33, 1608–1618. [Google Scholar] [CrossRef]
- Rose, C.; Parker, A.; Jefferson, B.; Cartmell, E. The characterization of feces and urine: A review of the literature to inform advanced treatment technology. Crit. Rev. Environ. Sci. Technol. 2015, 45, 1827–1879. [Google Scholar] [CrossRef]
- Case, L.P.; Daristotle, L.; Hayek, M.G.; Raasch, M.F. Canine and Feline Nutrition: A Resource for Companion Animal Professionals; Mosby Elsevier: Amsterdam, The Netherlands, 2011; ISBN 0323071473. [Google Scholar]
- Burrows, C.F.; Kronfeld, D.S.; Banta, C.A.; Merritt, A.M. Effects of fiber on digestibility and transit time in dogs. J. Nutr. 1982, 112, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.S.; Grieshop, C.M.; Clapper, G.M.; Shields, R.G.; Belay, T.; Merchen, N.R.; Fahey, G.C. Fruit and vegetable fiber fermentation by gut microflora from canines. J. Anim. Sci. 2001, 79, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Leone, V.; Gibbons, S.M.; Martinez, K.; Hutchison, A.L.; Huang, E.Y.; Cham, C.M.; Pierre, J.F.; Heneghan, A.F.; Nadimpalli, A.; Hubert, N.; et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe 2015, 17, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Bushman, F.D.; FitzGerald, G.A. Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc. Natl. Acad. Sci. USA 2015, 112, 10479–10484. [Google Scholar] [CrossRef]
- Smith, L.; Villaret-Cazadamont, J.; Claus, S.P.; Canlet, C.; Guillou, H.; Cabaton, N.J.; Ellero-Simatos, S. Important considerations for sample collection in metabolomics studies with a special focus on applications to liver functions. Metabolites 2020, 10. [Google Scholar] [CrossRef]
- Lamichhane, S.; Sundekilde, U.K.; Blædel, T.; Dalsgaard, T.K.; Larsen, L.H.; Dragsted, L.O.; Astrup, A.; Bertram, H.C. Optimizing sampling strategies for NMR-based metabolomics of human feces: Pooled vs. unpooled analyses. Anal. Methods 2017, 9, 4476–4480. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Depth Subsamples (n = 12) | Longitudinal Subsamples (n = 12) | ANOVA p-Value | ||||||
|---|---|---|---|---|---|---|---|---|
| Item | Inner | Outer | Cranial | Centre | Caudal | Depth Sub. | Longitudinal Sub. | Depth × Longitudinal Sub. |
| acetate (µmol/g) | 79.0 ± 20.0 | 60.2 ± 12.8 | 70.5 ± 20.9 | 69.8 ± 18.6 | 68.5 ± 19.6 | 0.002 | 0.954 | 0.725 |
| propionate (µmol/g) | 32.5 ± 16.8 | 20.7 ± 10.4 | 26.9 ± 15.1 | 24.8 ± 13.5 | 28.0 ± 17.6 | 0.007 | 0.809 | 0.834 |
| butyrate (µmol/g) | 21.0 ± 4.85 | 11.4 ± 3.03 | 15.3 ± 4.58 | 15.5 ± 6.13 | 17.7 ± 8.11 | 0.000 | 0.363 | 0.284 |
| isobutyrate (µmol/g) | 2.83 ± 1.11 | 2.09 ± 0.72 | 2.29 ± 0.74 | 2.57 ± 0.99 | 2.51 ± 1.27 | 0.058 | 0.815 | 0.930 |
| isovalerate (µmol/g) | 3.89 ± 2.08 | 2.74 ± 1.34 | 2.86 ± 1.31 | 3.50 ± 1.87 | 3.60 ± 2.25 | 0.099 | 0.627 | 0.916 |
| total SCFA (µmol/g) | 139.5 ± 37.9 | 97.2 ± 22.9 | 118.2 ± 38.9 | 116.3 ± 34.0 | 120.6 ± 43.2 | 0.000 | 0.932 | 0.646 |
| ammonia (µmol/g) | 46.7 ± 18.0 | 30.8 ± 8.81 | 35.7 ± 17.0 | 39.7 ± 14.2 | 40.9 ± 18.1 | 0.003 | 0.659 | 0.694 |
| pH | 6.54 ± 0.31 | 6.99 ± 0.32 | 6.69 ± 0.44 | 6.87 ± 0.33 | 6.73 ± 0.40 | 0.001 | 0.447 | 0.795 |
| Firmicutes (log10 copies/g) | 7.86 ± 0.85 | 7.74 ± 0.78 | 8.07 ± 0.72 | 7.74 ± 0.78 | 7.59 ± 0.90 | 0.667 | 0.353 | 0.907 |
| Bacteroidetes (log10 copies/g) | 7.57 ± 2.63 | 7.80 ± 2.72 | 7.77 ± 2.78 | 7.71 ± 2.80 | 7.58 ± 2.59 | 0.705 | 0.965 | 0.996 |
| Clostridium cluster I (log10 copies/g) | 7.40 ± 1.15 | 7.40 ± 1.49 | 7.67 ± 1.42 | 7.25 ± 1.39 | 7.27 ± 1.20 | 0.995 | 0.764 | 0.970 |
| Bifidobacterium spp. (log10 copies/g) | 3.14 ± 1.03 | 3.14 ± 1.00 | 3.22 ± 0.54 | 2.76 ± 1.50 | 3.42 ± 0.66 | 1.000 | 0.385 | 0.896 |
| Enterococcus spp. (log10 copies/g) | 6.68 ± 1.09 | 6.80 ± 1.03 | 6.86 ± 0.87 | 6.77 ± 1.15 | 6.60 ± 1.19 | 0.752 | 0.865 | 0.965 |
| Lactobacillus spp. (log10 copies/g) | 3.94 ± 0.82 | 3.86 ± 0.96 | 4.15 ± 0.95 | 3.80 ± 0.84 | 3.75 ± 0.88 | 0.789 | 0.448 | 0.677 |
| Item | Homogenised | Cranial Inner | Cranial Outer | Centre Inner | Centre Outer | Caudal Inner | Caudal Outer | ANOVA p-Value | Levene’s p-Value | |
|---|---|---|---|---|---|---|---|---|---|---|
| acetate (µmol/g) | mean ± SD | 75.2 ± 17.0 | 77.2 ± 23.6 | 63.7 ± 17.8 | 79.3 ± 21.0 | 60.4 ± 10.9 | 80.5 ± 20.0 | 56.5 ± 10.1 | 0.200 | |
| variance | 288.6 | 555.4 | 316.9 | 440.3 | 119.2 | 401.0 | 101.0 | 0.596 | ||
| propionate (µmol/g) | mean ± SD | 27.1 ± 14.1 | 32.0 ± 16.9 | 21.9 ± 12.7 | 29.9 ± 16.3 | 19.8 ± 8.96 | 35.6 ± 20.4 | 20.4 ± 11.5 | 0.531 | |
| variance | 200.1 | 284.1 | 161.6 | 265.2 | 80.2 | 415.0 | 133.1 | 0.701 | ||
| butyrate (µmol/g) | mean ± SD | 16.9 ± 3.30 a | 18.8 ± 3.16 a | 11.8 ± 2.52 a | 20.1 ± 4.72 a | 11.0 ± 3.24 a | 24.1 ± 5.63 b | 11.3 ± 3.83 a | <0.0001 | |
| variance | 10.9 | 10.0 | 6.37 | 22.3 | 10.5 | 31.7 | 14.7 | 0.074 | ||
| isobutyrate (µmol/g) | mean ± SD | 2.48 ± 0.87 | 2.60 ± 0.55 | 1.98 ± 0.83 | 2.90 ± 1.12 | 2.24 ± 0.81 | 2.98 ± 1.63 | 2.04 ± 0.65 | 0.567 | |
| variance | 0.76 | 0.30 | 0.69 | 1.26 | 0.65 | 2.65 | 0.42 | 0.755 | ||
| isovalerate (µmol/g) | mean ± SD | 3.38 ± 1.70 | 3.31 ± 1.20 | 2.41 ± 1.38 | 4.00 ± 2.12 | 3.00 ± 1.67 | 4.37 ± 2.92 | 2.82 ± 1.17 | 0.664 | |
| variance | 2.88 | 1.43 | 1.90 | 4.48 | 2.78 | 8.52 | 1.38 | 0.734 | ||
| total SCFA (µmol/g) | mean ± SD | 125.0 ± 29.8 | 134.6 ± 41.1 | 101.8 ± 32.3 | 136.2 ± 36.4 | 96.4 ± 16.9 | 147.8 ± 43.6 | 93.5 ± 21.4 | 0.073 | |
| variance | 886.5 | 1686.0 | 1045.4 | 1323.4 | 285.5 | 1898.3 | 458.0 | 0.399 | ||
| ammonia (µmol/g) | mean ± SD | 40.2 ± 13.2 | 41.9 ± 20.3 | 29.4 ± 11.9 | 46.4 ± 15.7 | 32.9 ± 9.70 | 51.8 ± 20.2 | 30.0 ± 5.19 | 0.158 | |
| variance | 175.0 | 412.1 | 142.1 | 245.2 | 94.0 | 409.8 | 26.9 | 0.721 | ||
| pH | mean ± SD | 6.79 ± 0.25 | 6.50 ± 0.39 | 6.87 ± 0.45 | 6.66 ± 0.31 | 7.08 ± 0.19 | 6.45 ± 0.25 | 7.02 ± 0.32 | 0.025 | |
| variance | 0.06 | 0.15 | 0.20 | 0.10 | 0.04 | 0.06 | 0.10 | 0.284 | ||
| Firmicutes (log10 copies/g) | mean ± SD | 7.84 ± 0.71 | 8.14 ± 0.80 | 7.99 ± 0.72 | 7.87 ± 0.78 | 7.62 ± 0.85 | 7.57 ± 1.03 | 7.61 ± 0.87 | 0.905 | |
| variance | 0.50 | 0.63 | 0.53 | 0.61 | 0.72 | 1.05 | 0.76 | 0.286 | ||
| Bacteroidetes (log10 copies/g) | mean ± SD | 7.67 ± 2.93 | 7.65 ± 2.96 | 7.89 ± 2.94 | 7.57 ± 2.86 | 7.85 ± 3.08 | 7.50 ± 2.70 | 7.66 ± 2.80 | 1.000 | |
| variance | 8.59 | 8.77 | 8.64 | 8.17 | 9.47 | 7.30 | 7.83 | 0.957 | ||
| Clostridium cluster I (log10 copies/g) | mean ± SD | 7.52 ± 1.17 | 7.66 ± 1.25 | 7.69 ± 1.71 | 7.19 ± 1.42 | 7.32 ± 1.52 | 7.35 ± 0.93 | 7.18 ± 1.54 | 0.994 | |
| variance | 1.37 | 1.57 | 2.94 | 2.03 | 2.31 | 0.86 | 2.38 | 0.388 | ||
| Bifidobacterium spp. (log10 copies/g) | mean ± SD | 3.27 ±0.47 | 3.12 ± 0.50 | 3.32 ± 0.61 | 2.75 ± 1.61 | 2.78 ± 1.58 | 3.54 ± 0.70 | 3.30 ± 0.66 | 0.843 | |
| variance | 0.22 | 0.25 | 0.37 | 2.59 | 2.48 | 0.49 | 0.44 | 0.301 | ||
| Enterococcus spp. (log10 copies/g) | mean ± SD | 6.92 ±1.14 | 6.83 ± 0.81 | 6.88 ± 1.02 | 6.75 ± 1.21 | 6.79 ± 1.23 | 6.47 ± 1.40 | 6.74 ± 1.08 | 0.997 | |
| variance | 1.30 | 0.66 | 1.05 | 1.47 | 1.52 | 1.97 | 1.17 | 0.895 | ||
| Lactobacillus spp. (log10 copies/g) | mean ± SD | 4.16 ±0.87 | 4.11 ± 0.94 | 4.20 ± 1.06 | 3.74 ± 1.00 | 3.85 ± 0.76 | 3.97 ± 0.62 | 3.54 ± 1.12 | 0.907 | |
| variance | 0.75 | 0.89 | 1.12 | 1.00 | 0.58 | 0.39 | 1.25 | 0.948 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinna, C.; Vecchiato, C.G.; Delsante, C.; Grandi, M.; Biagi, G. On the Variability of Microbial Populations and Bacterial Metabolites within the Canine Stool. An in-Depth Analysis. Animals 2021, 11, 225. https://doi.org/10.3390/ani11010225
Pinna C, Vecchiato CG, Delsante C, Grandi M, Biagi G. On the Variability of Microbial Populations and Bacterial Metabolites within the Canine Stool. An in-Depth Analysis. Animals. 2021; 11(1):225. https://doi.org/10.3390/ani11010225
Chicago/Turabian StylePinna, Carlo, Carla Giuditta Vecchiato, Costanza Delsante, Monica Grandi, and Giacomo Biagi. 2021. "On the Variability of Microbial Populations and Bacterial Metabolites within the Canine Stool. An in-Depth Analysis" Animals 11, no. 1: 225. https://doi.org/10.3390/ani11010225
APA StylePinna, C., Vecchiato, C. G., Delsante, C., Grandi, M., & Biagi, G. (2021). On the Variability of Microbial Populations and Bacterial Metabolites within the Canine Stool. An in-Depth Analysis. Animals, 11(1), 225. https://doi.org/10.3390/ani11010225