MHC Genotyping by SSCP and Amplicon-Based NGS Approach in Chamois


 ,
,  and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. MHC Genotyping by SSCP/Sanger Sequencing
2.2. MHC Genotyping by An Amplicon-Based NGS Approach
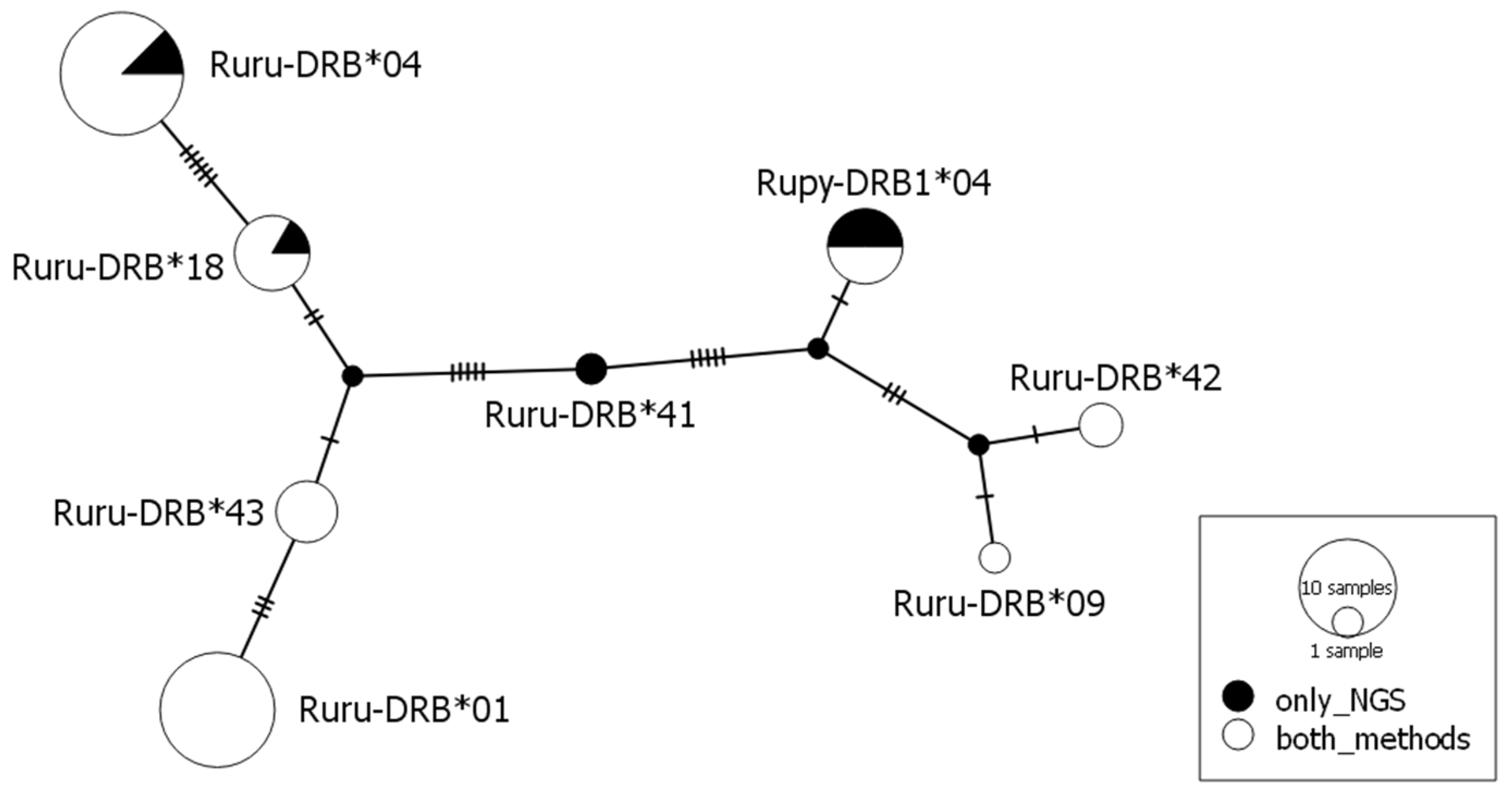
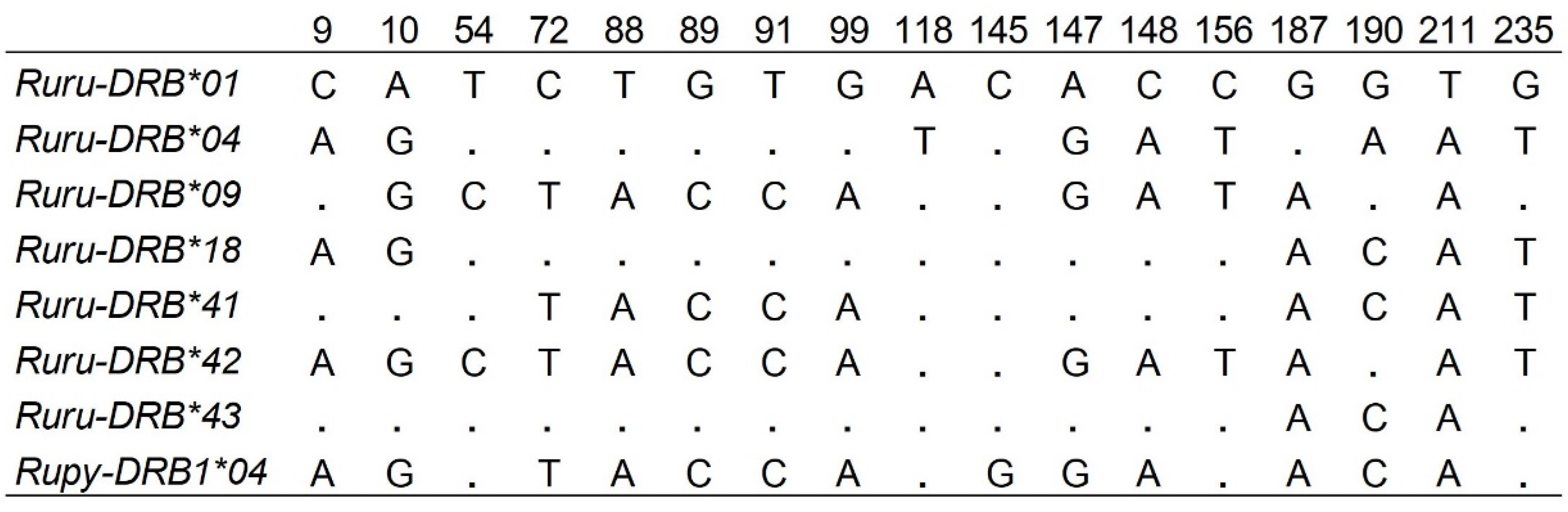
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leroy, G.; Carroll, E.L.; Bruford, M.W.; DeWoody, J.A.; Strand, A.; Waits, L.; Wang, J. Next-generation metrics for monitoring genetic erosion within populations of conservation concern. Evol. Appl. 2018, 11, 1066–1083. [Google Scholar] [CrossRef]
- Eizaguirre, C.; Baltazar-Soares, M. Evolutionary conservation-evaluating the adaptive potential of species. Evol. Appl. 2014, 7, 963–967. [Google Scholar] [CrossRef]
- Funk, W.C.; McKay, J.K.; Hohenlohe, P.A.; Allendorf, F.W. Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 2012, 27, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2005, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Santillán, D.D.; Lacey, E.A.; Gendron, D.; Ortega, J. Genetic variation at exon 2 of the MHC class II DQB locus in blue whale (Balaenoptera musculus) from the Gulf of California. PLoS ONE 2016, 11, 1–15. [Google Scholar] [CrossRef]
- Spurgin, L.G.; Richardson, D.S. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc. R. Soc. B Biol. Sci. 2010, 277, 979–988. [Google Scholar] [CrossRef]
- O’Connor, E.A.; Strandh, M.; Hasselquist, D.; Nilsson, J.Å.; Westerdahl, H. The evolution of highly variable immunity genes across a passerine bird radiation. Mol. Ecol. 2016, 25, 977–989. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-Throughput Sequencing Technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef]
- Babik, W.; Taberlet, P.; Ejsmond, M.J.; Radwan, J. New generation sequencers as a tool for genotyping of highly polymorphic multilocus MHC system. Mol. Ecol. Resour. 2009, 9, 713–719. [Google Scholar] [CrossRef]
- Zagalska-Neubauer, M.; Babik, W.; Stuglik, M.; Gustafsson, L.; Cichoń, M.; Radwan, J. 454 sequencing reveals extreme complexity of the class II Major Histocompatibility Complex in the collared flycatcher. BMC Evol. Biol. 2010, 10, 395. [Google Scholar] [CrossRef]
- Lighten, J.; van Oosterhout, C.; Paterson, I.G.; McMullan, M.; Bentzen, P. Ultra-deep Illumina sequencing accurately identifies MHC class IIb alleles and provides evidence for copy number variation in the guppy (Poecilia reticulata). Mol. Ecol. Resour. 2014, 14, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Grogan, K.E.; McGinnis, G.J.; Sauther, M.L.; Cuozzo, F.P.; Drea, C.M. Next-generation genotyping of hypervariable loci in many individuals of a non-model species: Technical and theoretical implications. BMC Genomics 2016, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Zhang, P.; Dunn, D.W.; Wang, T.; Mi, R.; Li, B. Assigning alleles to different loci in amplifications of duplicated loci. Mol. Ecol. Resour. 2019, 19, 1240–1253. [Google Scholar] [CrossRef] [PubMed]
- Lighten, J.; van Oosterhout, C.; Bentzen, P. Critical review of NGS analyses for de novo genotyping multigene families. Mol. Ecol. 2014, 23, 3957–3972. [Google Scholar] [CrossRef] [PubMed]
- Biedrzycka, A.; Sebastian, A.; Migalska, M.; Westerdahl, H.; Radwan, J. Testing genotyping strategies for ultra-deep sequencing of a co-amplifying gene family: MHC class I in a passerine bird. Mol. Ecol. Resour. 2017, 17, 642–655. [Google Scholar] [CrossRef]
- Rekdal, S.L.; Anmarkrud, J.A.; Johnsen, A.; Lifjeld, J.T. Genotyping strategy matters when analyzing hypervariable major histocompatibility complex-Experience from a passerine bird. Ecol. Evol. 2018, 8, 1680–1692. [Google Scholar] [CrossRef]
- Babik, W. Methods for MHC genotyping in non-model vertebrates. Mol. Ecol. Resour. 2010, 10, 237–251. [Google Scholar] [CrossRef]
- Sunnucks, P.; Wilson, A.C.C.; Beheregaray, L.B.; Zenger, K.; French, J.; Taylor, A.C. SSCP is not so difficult: The application and utility of single-stranded conformation polymorphism in evolutionary biology and molecular ecology. Mol. Ecol. 2000, 9, 1699–1710. [Google Scholar] [CrossRef]
- Garrigan, D.; Hedrick, P.W. Class I MHC polymorphism and evolution in endangered California Chinook and other Pacific salmon. Immunogenetics 2001, 53, 483–489. [Google Scholar] [CrossRef]
- Noakes, M.A.; Reimer, T.; Phillips, R.B. Genotypic characterization of an MHC class II locus in lake trout (Salvelinus namaycush) from Lake Superior by single-stranded conformational polymorphism analysis and reference strand-mediated conformational analysis. Mar. Biotechnol. 2003, 5, 270–278. [Google Scholar] [CrossRef]
- Hughes, A.L.; Yeager, M. Natural selection at major histocompatibility complex loci of vertebrates. Annu. Rev. Genet. 1998, 32, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Schaschl, H.; Goodman, S.J.; Suchentrunk, F. Sequence analysis of the MHC class II DRB alleles in Alpine chamois (Rupicapra r. rupicapra). Dev. Comp. Immunol. 2004, 28, 265–277. [Google Scholar] [CrossRef]
- Číková, D.; de Bellocq, J.G.; Baird, S.J.E.; Piálek, J.; Bryja, J. Genetic structure and contrasting selection pattern at two major histocompatibility complex genes in wild house mouse populations. Heredity 2011, 106, 727–740. [Google Scholar] [CrossRef]
- Bryja, J.; Galan, M.; Charbonnel, N.; Cosson, J.F. Analysis of major histocompatibility complex class II gene in water voles using capillary electrophoresis-single stranded conformation polymorphism. Mol. Ecol. Notes 2005, 5, 173–176. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, A.; Herdegen, M.; Migalska, M.; Radwan, J. Amplisas: A web server for multilocus genotyping using next-generation amplicon sequencing data. Mol. Ecol. Resour. 2016, 16, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Fuselli, S.; Baptista, R.P.; Panziera, A.; Magi, A.; Guglielmi, S.; Tonin, R.; Benazzo, A.; Bauzer, L.G.; Mazzoni, C.J.; Bertorelle, G. A new hybrid approach for MHC genotyping: High-throughput NGS and long read MinION nanopore sequencing, with application to the non-model vertebrate Alpine chamois (Rupicapra rupicapra). Heredity 2018, 121, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Zemanová, B.; Hájková, P.; Hájek, B.; Martínková, N.; Mikulíček, P.; Zima, J.; Bryja, J. Extremely low genetic variation in endangered Tatra chamois and evidence for hybridization with an introduced Alpine population. Conserv. Genet. 2015, 16, 729–741. [Google Scholar] [CrossRef]
- Mona, S.; Crestanello, B.; Bankhead-Dronnet, S.; Pecchioli, E.; Ingrosso, S.; D’Amelio, S.; Rossi, L.; Meneguz, P.G.; Bertorelle, G. Disentangling the effects of recombination, selection, and demography on the genetic variation at a major histocompatibility complex class II gene in the alpine chamois. Mol. Ecol. 2008, 17, 4053–4067. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Schaschl, H.; Wandeler, P.; Suchentrunk, F.; Obexer-Ruff, G.; Goodman, S.J. Selection and recombination drive the evolution of MHC class II DRB diversity in ungulates. Heredity 2006, 97, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Busto, J.; García-Etxebarria, K.; Herrero, J.; Garin, I.; Jugo, B.M. Diversity and evolution of the Mhc-DRB1 gene in the two endemic Iberian subspecies of Pyrenean chamois, Rupicapra pyrenaica. Heredity 2007, 99, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S.; Courtiol, A.; Mazzoni, C.J. MHC genotyping of non-model organisms using next-generation sequencing: A new methodology to deal with artefacts and allelic dropout. BMC Genomics 2013, 14, 542. [Google Scholar] [CrossRef] [PubMed]
- Montero, B.K.; Refaly, E.; Ramanamanjato, J.B.; Randriatafika, F.; Rakotondranary, S.J.; Wilhelm, K.; Ganzhorn, J.U.; Sommer, S. Challenges of next-generation sequencing in conservation management: Insights from long-term monitoring of corridor effects on the genetic diversity of mouse lemurs in a fragmented landscape. Evol. Appl. 2019, 12, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Radwan, J.; Zagalska-Neubauer, M.; Cichoń, M.; Sendecka, J.; Kulma, K.; Gustafsson, L.; Babik, W. MHC diversity, malaria and lifetime reproductive success in collared flycatchers. Mol. Ecol. 2012, 21, 2469–2479. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Allele | SSCP/Sanger | NGS | ||
|---|---|---|---|---|
| No Observations | Allele Frequency | No Observations | Allele Frequency | |
| Ruru-DRB*01 | 20 | 0.357 | 18 | 0.321 |
| Ruru-DRB*04 | 18 | 0.321 | 17 | 0.304 |
| Ruru-DRB*09 | 1 | 0.018 | 1 | 0.018 |
| Ruru-DRB*18 | 5 | 0.089 | 6 | 0.107 |
| Ruru-DRB*41 | 0 | 0.000 | 1 | 0.018 |
| Ruru-DRB*42 | 2 | 0.036 | 2 | 0.036 |
| Ruru-DRB*43 | 6 | 0.107 | 4 | 0.071 |
| Rupy-DRB1*04 | 4 | 0.071 | 7 | 0.125 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stipoljev, S.; Bužan, E.; Rolečková, B.; Iacolina, L.; Šprem, N. MHC Genotyping by SSCP and Amplicon-Based NGS Approach in Chamois. Animals 2020, 10, 1694. https://doi.org/10.3390/ani10091694
Stipoljev S, Bužan E, Rolečková B, Iacolina L, Šprem N. MHC Genotyping by SSCP and Amplicon-Based NGS Approach in Chamois. Animals. 2020; 10(9):1694. https://doi.org/10.3390/ani10091694
Chicago/Turabian StyleStipoljev, Sunčica, Elena Bužan, Barbora Rolečková, Laura Iacolina, and Nikica Šprem. 2020. "MHC Genotyping by SSCP and Amplicon-Based NGS Approach in Chamois" Animals 10, no. 9: 1694. https://doi.org/10.3390/ani10091694
APA StyleStipoljev, S., Bužan, E., Rolečková, B., Iacolina, L., & Šprem, N. (2020). MHC Genotyping by SSCP and Amplicon-Based NGS Approach in Chamois. Animals, 10(9), 1694. https://doi.org/10.3390/ani10091694