Optimization of Sperm Cryopreservation Protocol for Peregrine Falcon (Falco peregrinus)

 ,
,  , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Semen Collection
2.3. Experimental Design
2.4. Sperm Evaluation
2.4.1. Sperm Motility
2.4.2. Sperm Viability and Acrosome Status
2.4.3. Mitochondrial Status
2.4.4. DNA Fragmentation
2.5. Statistical Analysis
3. Results
3.1. Experiment 1: Effect of the Freezing Method and Thawing Conditions on Sperm Quality
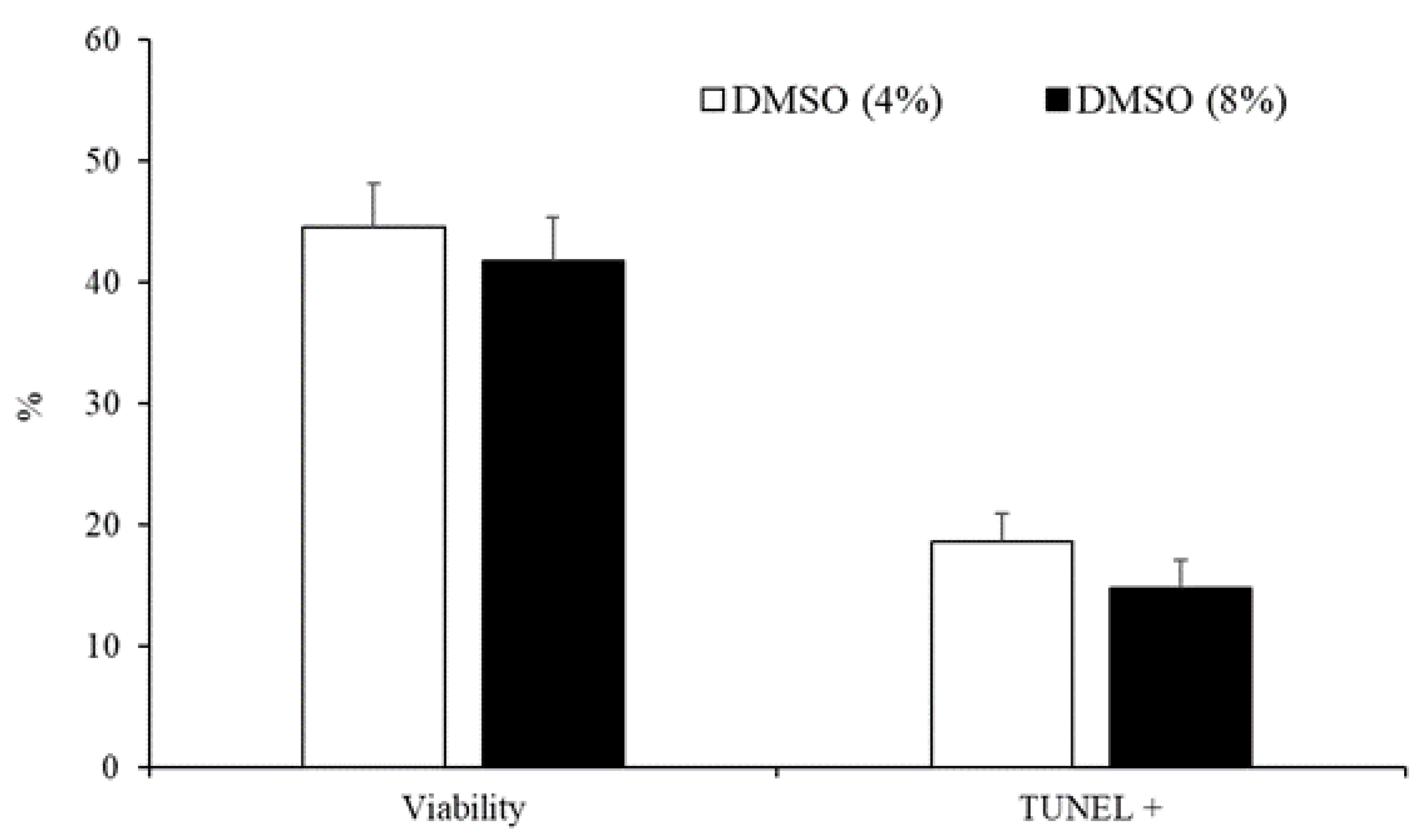
3.2. Experiment 2: Effect of the Type and Concentration of Cryoprotectant on Sperm Quality
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The IUCN Red List of Threatened Species. Available online: https://www.iucnredlist.org/ (accessed on 30 May 2019).
- Pukazhenthi, B.S.; Wildt, D.E. Which reproductive technologies are most relevant to studying, managing and conserving wildlife? Reprod. Fertil. Dev. 2004, 16, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Prieto, M.T.; Sánchez-Calabuig, M.J.; Hildebrandt, T.B.; Saragusty, J. Sperm cryopreservation in wild animals. Eur J. Wildl. Res. 2014, 60, 851–864. [Google Scholar] [CrossRef]
- Long, J. Successful cryopreservation of avian germplasm: Why a multifaceted approach is required. Cryobiology 2013, 67, 406. [Google Scholar] [CrossRef]
- Rosen, M.; Marsh, P.; Quinn, M. Gamete and Embryo Manipulation. In Yen and Jaffe’s Reproductive Endocrinology, 8th ed.; Strauss, J.F., Barbieri, R.L., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; pp. 823–856. [Google Scholar]
- FAO. Cryoconservation of Animal Genetic Resources; FAO: Rome, Italy, 2012. [Google Scholar]
- Brock, M.; Bird, D.; Ansah, G. Cryogenic preservation of spermatozoa of the American kestrel. Int. Zoo Yearb. 1983, 23, 67–71. [Google Scholar] [CrossRef]
- Villaverde-Morcillo, S.; Soler, A.J.; Esteso, M.C.; Castaño, C.; Gonzalez, F. Immature and mature sperm morphometry in fresh and frozen-thawed falcon ejaculates. Theriogenology 2017, 98, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.A.; Lierz, M. Veterinary Aspects of Bird of Prey Reproduction. Vet. Clin. N. Am. Exot. Anim. Pract. 2017, 20, 455–483. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, M.; Berlinguer, F.; Ledda, M.; Leoni, G.G.; Satta, V.; Succu, S.; Rotta, A.; Pasciu, V.; Zinellu, A.; Carru, C.; et al. Ejaculate collection efficiency and post-thaw semen quality in wild-caught Griffon vultures from the Sardinian population. Reprod. Biol. Endocrinol. 2009, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Long, J.A. Avian Semen Cryopreservation: What Are the Biological Challenges? Poult. Sci. 2006, 85, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Blesbois, E. Biological features of the avian male gamete and their application to biotechnology of conservation. J. Poult. Sci. 2012, 49, 141–149. [Google Scholar] [CrossRef]
- Al-Daraji, H.J.; Al-Shemmary, S.A. Effect of breed of falcon on semen quality traits. Int. J. Conserv. Sci. 2016, 7, 725–734. [Google Scholar]
- Mphaphathi, M.; Luseba, D.; Sutherland, B.; Nedambale, T. Comparison of slow freezing and vitrification methods for Venda cockerel’s spermatozoa. Open J. Anim. Sci. 2012, 2, 204–210. [Google Scholar] [CrossRef]
- Santiago-Moreno, J.; Castaño, C.; Toledano-Díaz, A.; Coloma, M.A.; López-Sebastián, A.; Prieto, M.T.; Campo, J.L. Semen cryopreservation for the creation of a Spanish poultry breeds cryobank: Optimization of freezing rate and equilibration time. Poult. Sci. 2011, 90, 2047–2053. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.M.; Long, J.A.; Gee, G.; Wildt, D.E.; Donoghue, A.M. Comparative cryopreservation of avian spermatozoa: Effects of freezing and thawing rates on turkey and sandhill crane sperm cryosurvival. Anim. Reprod. Sci. 2012, 131, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, A.M.; Wishart, G.J. Storage of poultry semen. Anim. Reprod. Sci. 2000, 62, 213–232. [Google Scholar] [CrossRef]
- Bóveda, P.; Toledano-Díaz, A.; Castaño, C.; Esteso, M.C.; López-Sebastian, A.; Rizos, D.; Bielli, A.; Ungerfeld, R.; Santiago-Moreno, J. Ultra-rapid cooling of ibex sperm by spheres method does not induce a vitreous extracellular state and increases the membrane damages. PLoS ONE 2020, 15, e0227946. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Moreno, J.; Castaño, C.; Toledano-Díaz, A.; Esteso, M.C.; Martínez-Nevado, E.; Gimeno-Martínez, J.; López-Goya, A. Semen cryopreservation in black-footed (Spheniscus demersus) and gentoo (Pygoscelis papua) penguins: Effects of thawing temperature on semen characteristics. Anim. Reprod. Sci. 2019, 200, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Abouelezz, F.M.; Castaño, C.; Toledano-Díaz, A.; Esteso, M.C.; López-Sebastián, A.; Campo, J.L.; Santiago-Moreno, J. Effect of the Interaction Between Cryoprotectant Concentration and Cryopreservation Method on Frozen/Thawed Chicken Sperm Variables. Reprod. Domest. Anim. 2015, 50, 135–141. [Google Scholar] [CrossRef]
- Iaffaldano, N.; Di Iorio, M.; Miranda, M.; Zaniboni, L.; Manchisi, A.; Cerolini, S. Cryopreserving turkey semen in straws and nitrogen vapour using DMSO or DMA: Effects of cryoprotectant concentration, freezing rate and thawing rate on post-thaw semen quality. Br. Poult. Sci. 2016, 57, 264–270. [Google Scholar] [CrossRef]
- Brown, M.E.; Singh, R.P.; Pukazhenthi, B.; Keefer, C.L.; Songsasen, N. Cryopreservation effects on sperm function and fertility in two threatened crane species. Cryobiology 2018, 82, 148–154. [Google Scholar] [CrossRef]
- Rakha, B.A.; Ansari, M.S.; Akhter, S.; Zafar, Z.; Naseer, A.; Hussain, I.; Santiago-Moreno, J.; Blesbois, E. Dimethyleacetamide improves the cryosurvivability of Indian red jungle fowl (Gallus gallus murghi) sperm. Theriogenology 2017, 103, 83–89. [Google Scholar] [CrossRef]
- Rakha, B.A.; Ansari, M.S.; Akhter, S.; Zafar, Z.; Naseer, A.; Hussain, I.; Blesbois, E.; Santiago-Moreno, J. Use of dimethylsulfoxide for semen cryopreservation in India red jungle fowl (Gallus gallus murghi). Theriogenology 2018, 122, 61–67. [Google Scholar] [CrossRef]
- Gurtovenko, A.A.; Anwar, J. Modulating the structure and properties of cell membranes: The molecular mechanism of action of dimethyl sulfoxide. J. Phys. Chem. B 2007, 111, 10453–10460. [Google Scholar] [CrossRef]
- Graham, E.F.; Nelson, D.S.; Schmehl, M.K. Development of extender and techniques for frozen turkey semen. Poult. Sci. 1982, 61, 550–557. [Google Scholar] [CrossRef]
- Herrera, J.A.; Quintana, J.A.; López, M.A.; Betancourt, M.; Fierro, R. Individual cryopreservation with dimethyl sulfoxide and polyvinylpyrrolidone of ejaculates and pooled semen of three avian species. Arch. Androl. 2005, 51, 353–360. [Google Scholar] [CrossRef]
- Blanco, J.M.; Long, J.A.; Gee, G.; Wildt, D.E.; Donoghue, A.M. Comparative cryopreservation of avian spermatozoa: Benefits of non-permeating osmoprotectants and ATP on turkey and crane sperm. Anim. Reprod. Sci. 2011, 123, 242–248. [Google Scholar] [CrossRef]
- Han, X.F.; Niu, Z.Y.; Liu, F.Z.; Yang, C.S. Effects of diluents, cryoprotectants, equilibration time and thawing temperature on cryopreservation of duck semen. Int. J. Poult. Sci. 2005, 4, 197–201. [Google Scholar]
- Gee, G.F.; Morrell, C.A.; Franson, J.C.; Pattee, O.H. Cryopreservation of American kestrel semen with dimethylsulfoxide. J. Raptor Res. 1993, 27, 21–25. [Google Scholar]
- Blanco, J.M.; Gee, G.; Wildt, D.E.; Donoghue, A.M. Species Variation in Osmotic, Cryoprotectant, and Cooling Rate Tolerance in Poultry, Eagle, and Peregrine Falcon Spermatozoa. Biol. Reprod. 2000, 63, 1164–1171. [Google Scholar] [CrossRef]
- Blanco, J.M.; Long, J.A.; Gee, G.; Donoghue, A.M.; Wildt, D.E. Osmotic tolerance of avian spermatozoa: Influence of time, temperature, cryoprotectant and membrane ion pump function on sperm viability. Cryobiology 2008, 56, 8–14. [Google Scholar] [CrossRef]
- Blesbois, E.; Grasseau, I.; Seigneurin, F. Membrane fluidity and the ability of domestic bird spermatozoa to survive cryopreservation. Reproduction 2004, 129, 371–378. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Concentration (× 106/mL) | 12.3 ± 1.3 |
| Total motility (%) | 73.8 ± 2.6 |
| Progressive motility (%) | 24.7 ± 4.0 |
| Straight-line Velocity (VSL; µm/s) | 27.0 ± 1.1 |
| Straightness (STR; %) | 82.6 ± 1.4 |
| Amplitude of Lateral Head (ALH; µm) | 2.1 ± 0.1 |
| Beat-cross frequency (BCF; Hz) | 9.8 ± 0.4 |
| Viability (%) | 82.3 ± 2.9 |
| TUNEL + (%) | 4.8 ± 1.5 |
| Semen Variables | DMA 3% | DMSO 8% |
|---|---|---|
| Motile spermatozoa (%) | 4.2 ± 1.9 a | 13.9 ± 1.8 b |
| Total motility (%) | 7.7 ± 2.3 a | 21.1 ± 2.3 b |
| Progressive motility (%) | 1.1 ± 0.3 a | 2.1 ± 0.3 b |
| Straight-line Velocity (VSL; µm/s) | 7.7 ± 1.4a | 12.9 ± 1.4b |
| Straightness (STR; %) | 49 ± 6.4 a | 70.6 ± 6.2 b |
| Amplitude of Lateral Head (ALH; µm) | 0.3 ± 0.1 a | 0.8 ± 0.1 b |
| Beat-cross frequency (BCF; Hz) | 1.2 ± 0.5 a | 3.1 ± 0.5 b |
| Semen Variables | DMSO 4% | DMSO 8% |
|---|---|---|
| Motile spermatozoa (%) | 9.5 ± 1.9 | 8.1 ± 1.9 |
| Total motility (%) | 14.5 ± 2.7 | 12.6 ± 2.7 |
| Progressive motility (%) | 2.0 ± 0.5 | 1.6 ± 0.5 |
| Straight-line Velocity (VSL; µm/s) | 16.1 ± 2.9 | 13.2 ± 2.9 |
| Straightness (STR; %) | 77.8 ± 10.4 | 69.4 ± 10.4 |
| Amplitude of Lateral Head (ALH; µm) | 0.4 ± 0.2 | 0.4 ± 0.2 |
| Beat-cross frequency (BCF; Hz) | 2.5 ± 0.8 | 2.5 ± 0.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, B.; Sánchez-Ajofrín, I.; Castaño, C.; García-Álvarez, O.; Esteso, M.C.; Maroto-Morales, A.; Iniesta-Cuerda, M.; Garde, J.J.; Santiago-Moreno, J.; Soler, A.J. Optimization of Sperm Cryopreservation Protocol for Peregrine Falcon (Falco peregrinus). Animals 2020, 10, 691. https://doi.org/10.3390/ani10040691
Cardoso B, Sánchez-Ajofrín I, Castaño C, García-Álvarez O, Esteso MC, Maroto-Morales A, Iniesta-Cuerda M, Garde JJ, Santiago-Moreno J, Soler AJ. Optimization of Sperm Cryopreservation Protocol for Peregrine Falcon (Falco peregrinus). Animals. 2020; 10(4):691. https://doi.org/10.3390/ani10040691
Chicago/Turabian StyleCardoso, Beatriz, Irene Sánchez-Ajofrín, Cristina Castaño, Olga García-Álvarez, Milagros Cristina Esteso, Alejandro Maroto-Morales, María Iniesta-Cuerda, José Julián Garde, Julián Santiago-Moreno, and Ana Josefa Soler. 2020. "Optimization of Sperm Cryopreservation Protocol for Peregrine Falcon (Falco peregrinus)" Animals 10, no. 4: 691. https://doi.org/10.3390/ani10040691
APA StyleCardoso, B., Sánchez-Ajofrín, I., Castaño, C., García-Álvarez, O., Esteso, M. C., Maroto-Morales, A., Iniesta-Cuerda, M., Garde, J. J., Santiago-Moreno, J., & Soler, A. J. (2020). Optimization of Sperm Cryopreservation Protocol for Peregrine Falcon (Falco peregrinus). Animals, 10(4), 691. https://doi.org/10.3390/ani10040691