
Anaerobic Fungal Mevalonate Pathway Genomic Biases Lead to Heterologous Toxicity Underpredicted by Codon Adaptation Indices

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Homolog Identification, Primer Design, PCR, RT-PCR, and Cloning
2.2. Growth Analysis
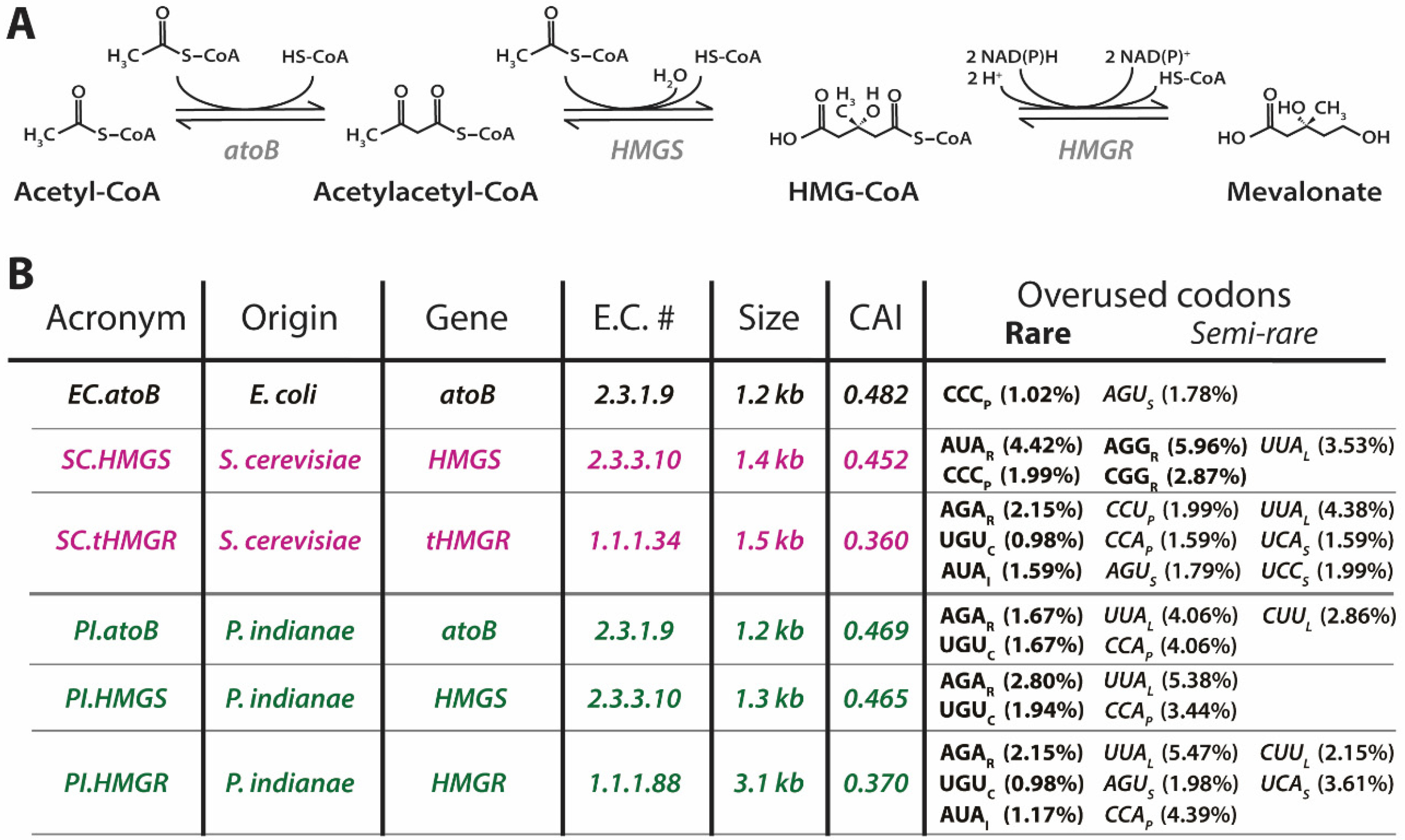
2.3. Codon Adaptation Index, Codon Usage, and Codon Optimization
2.4. Mevalonate Production Cultures and HPLC Analysis
3. Results and Discussion
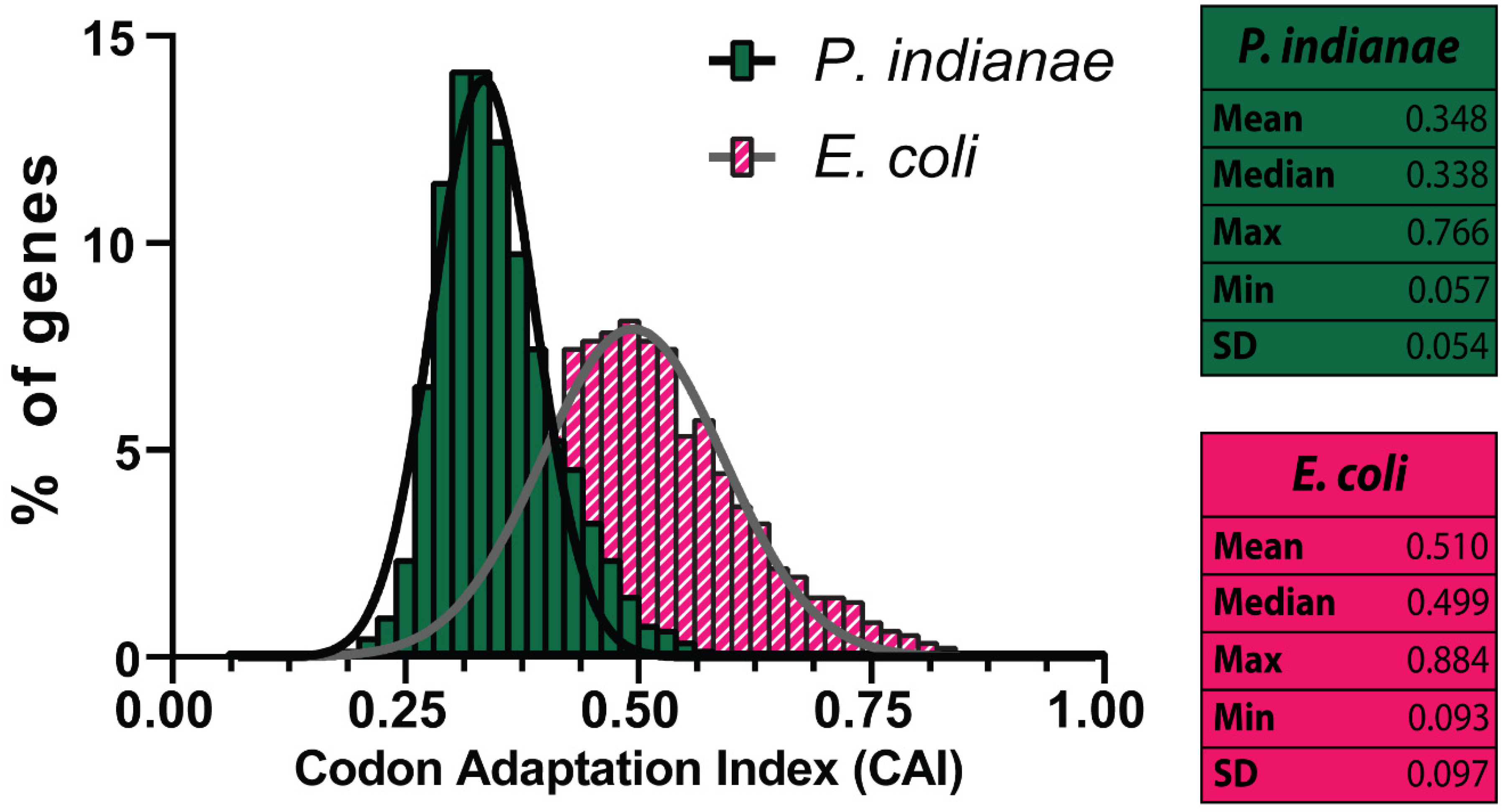
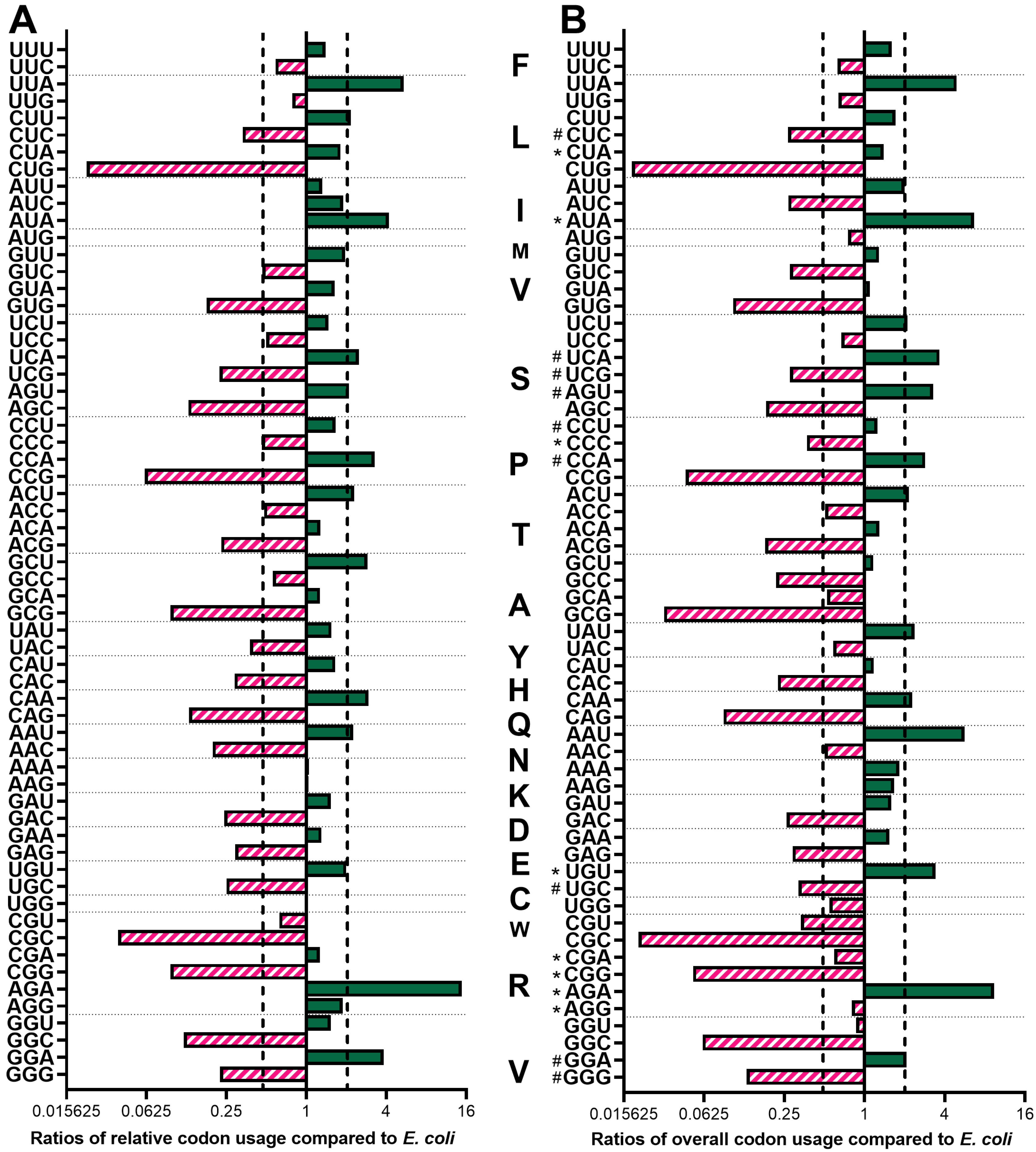
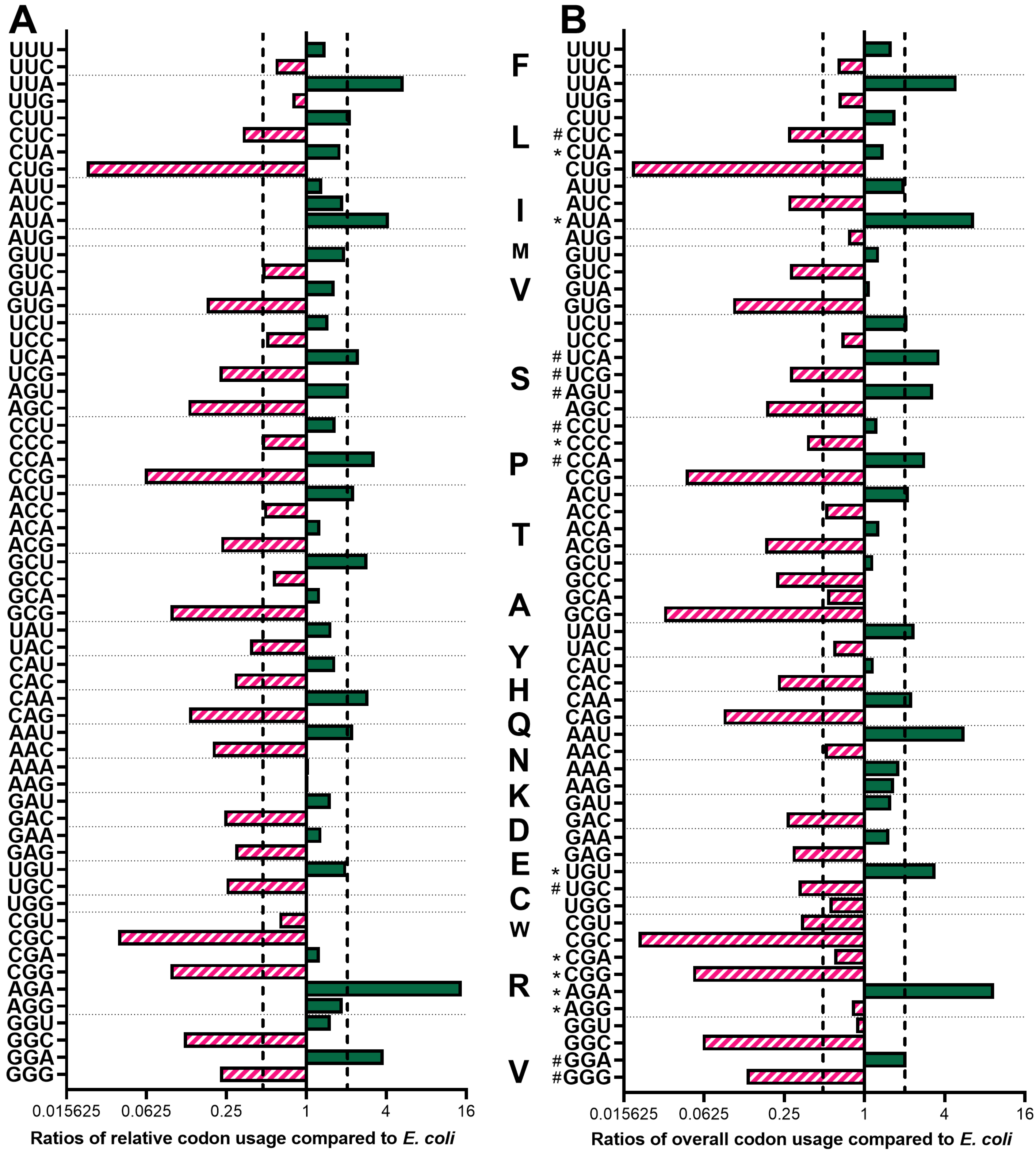
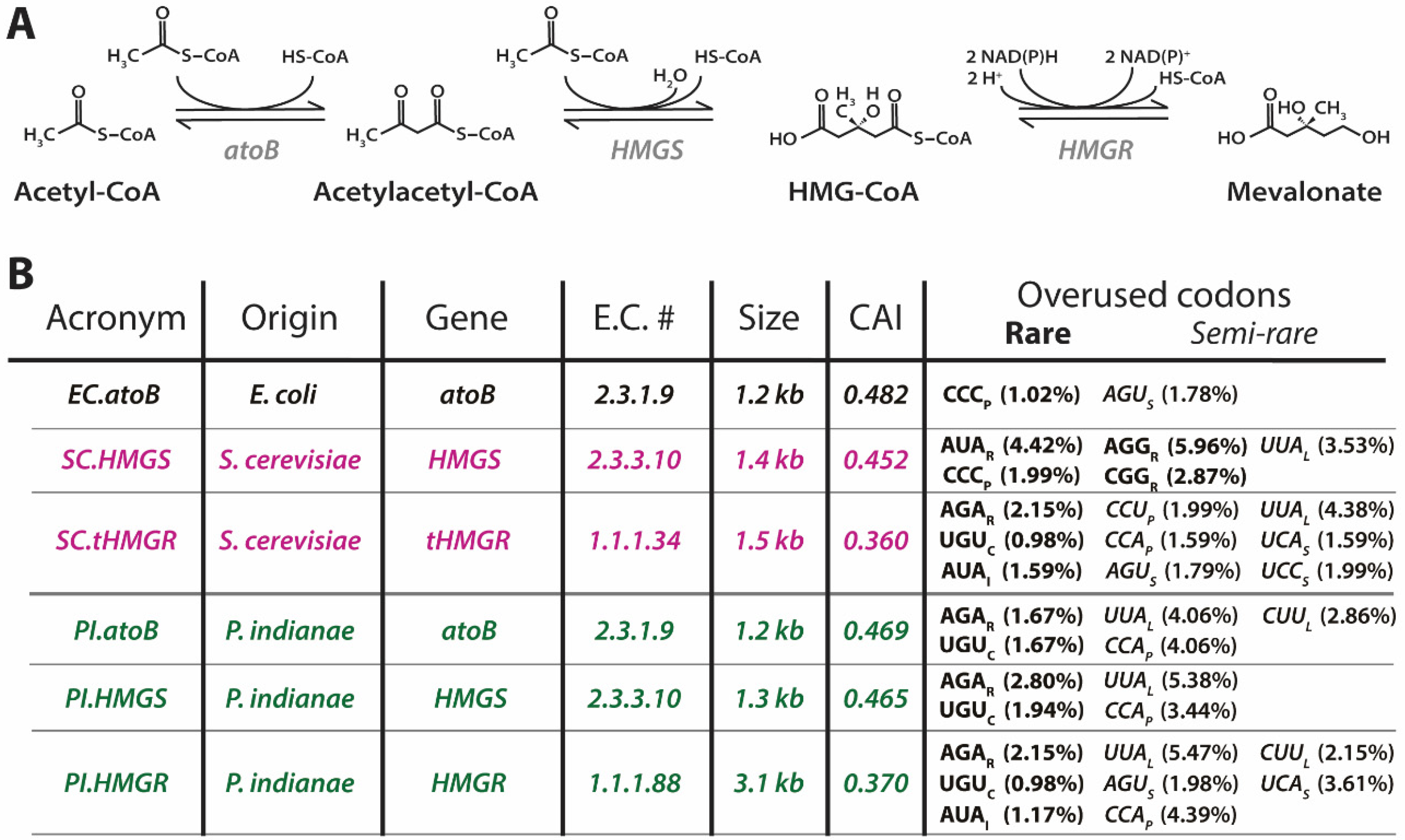
3.1. Codon Usage and Preferences of P. indianae Are Strongly AT-Biased
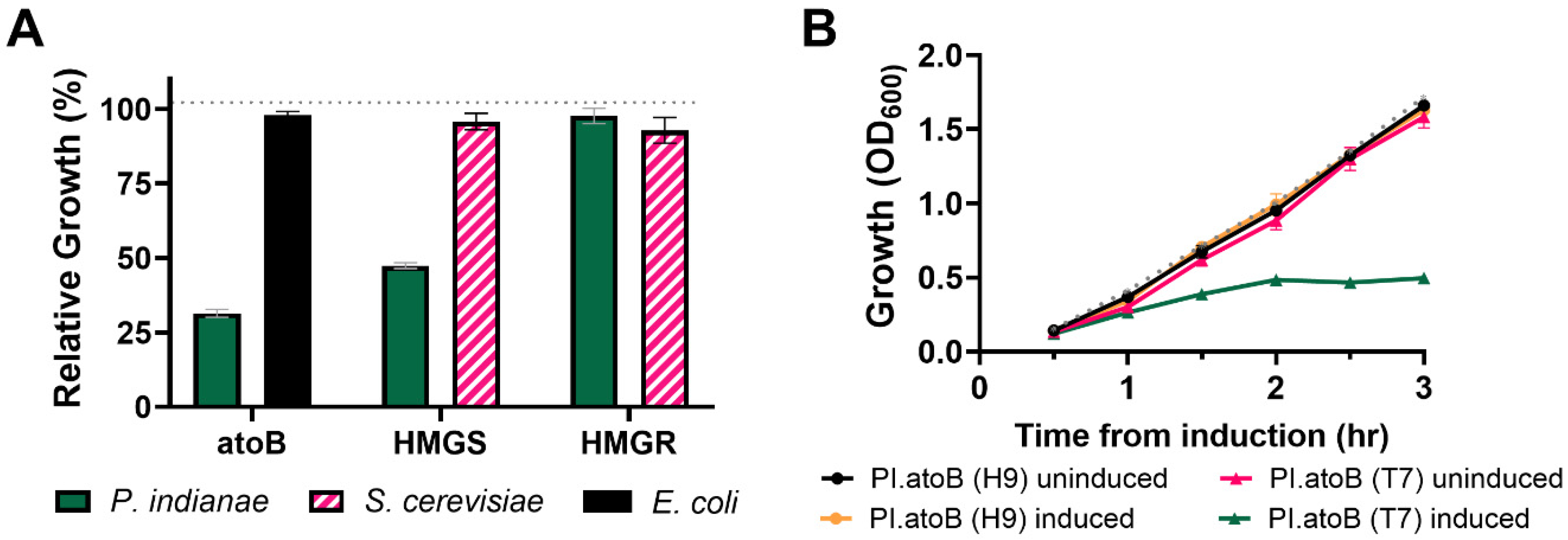
3.2. E.coli Are Not Well Equipped to Express the AT-rich Genes of P. indianae
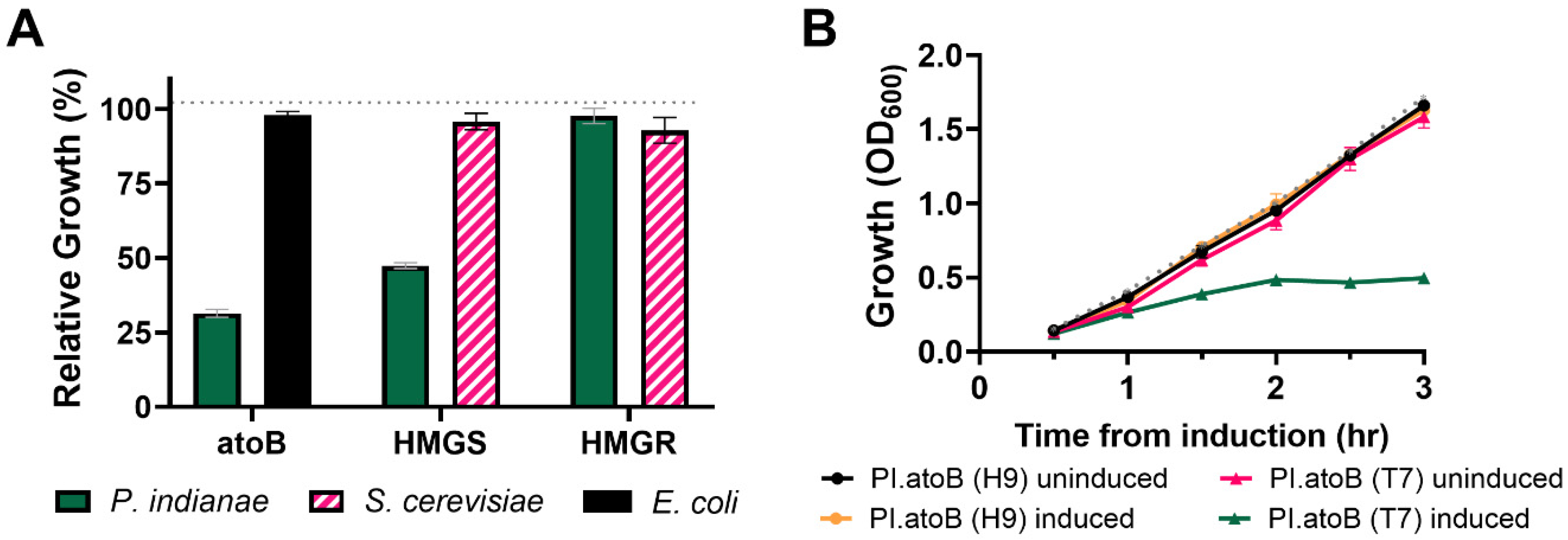
3.3. Strains with Additional tRNAs for Rare Codons Do Not Effectively Relieve the Burden of Expressing P. indianae Genes
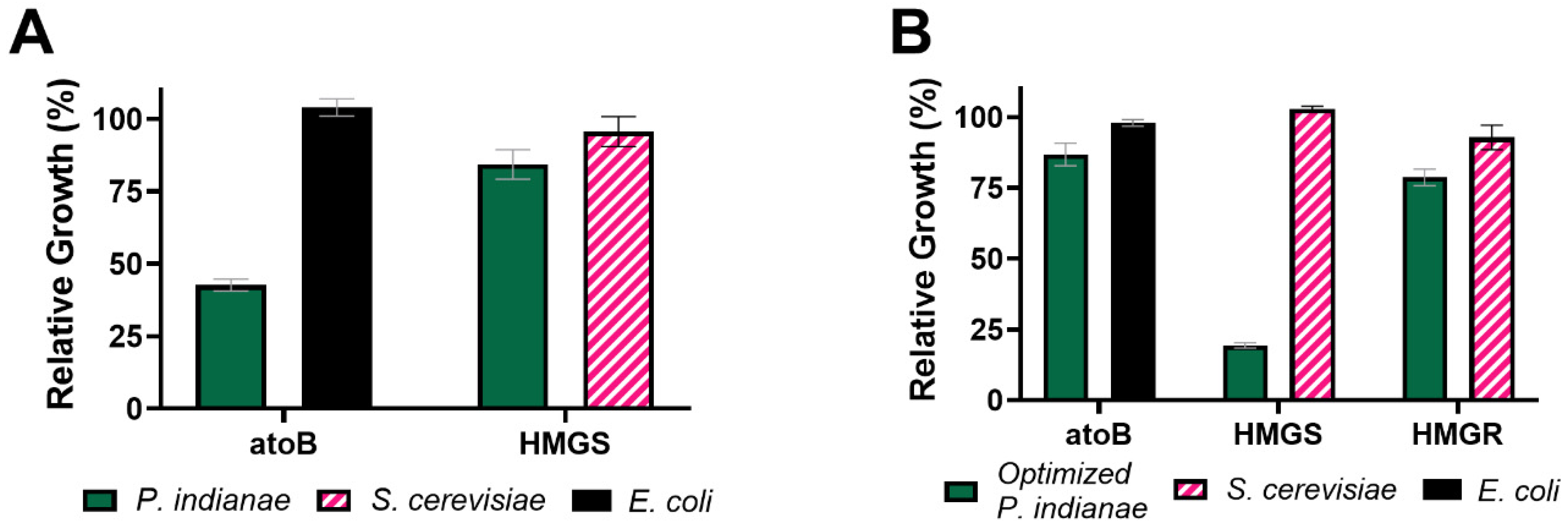
3.4. Codon Optimization Alleviates the Growth Deficiencies Seen When the P. indianae atoB Is Expressed in E. coli
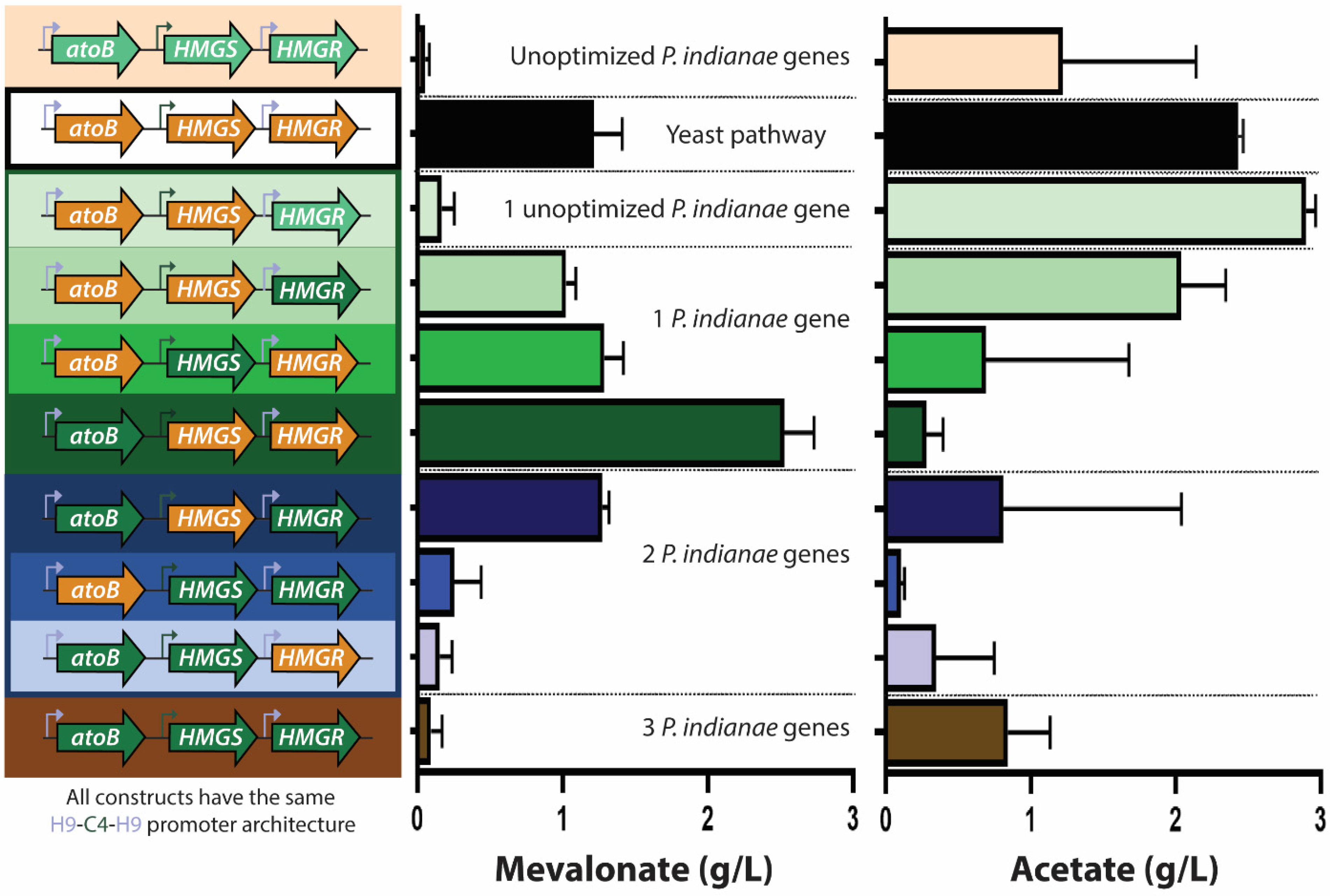
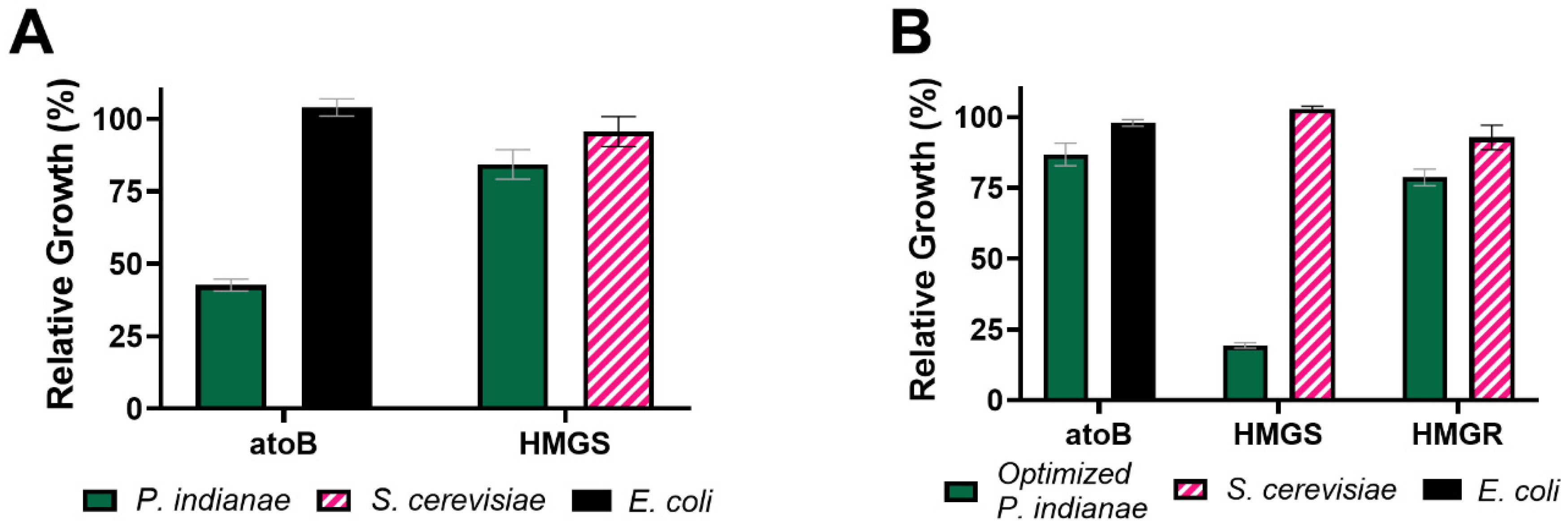
3.5. Expression of Unoptimized Genes Hinders Biosynthesis from P. indianae Genes
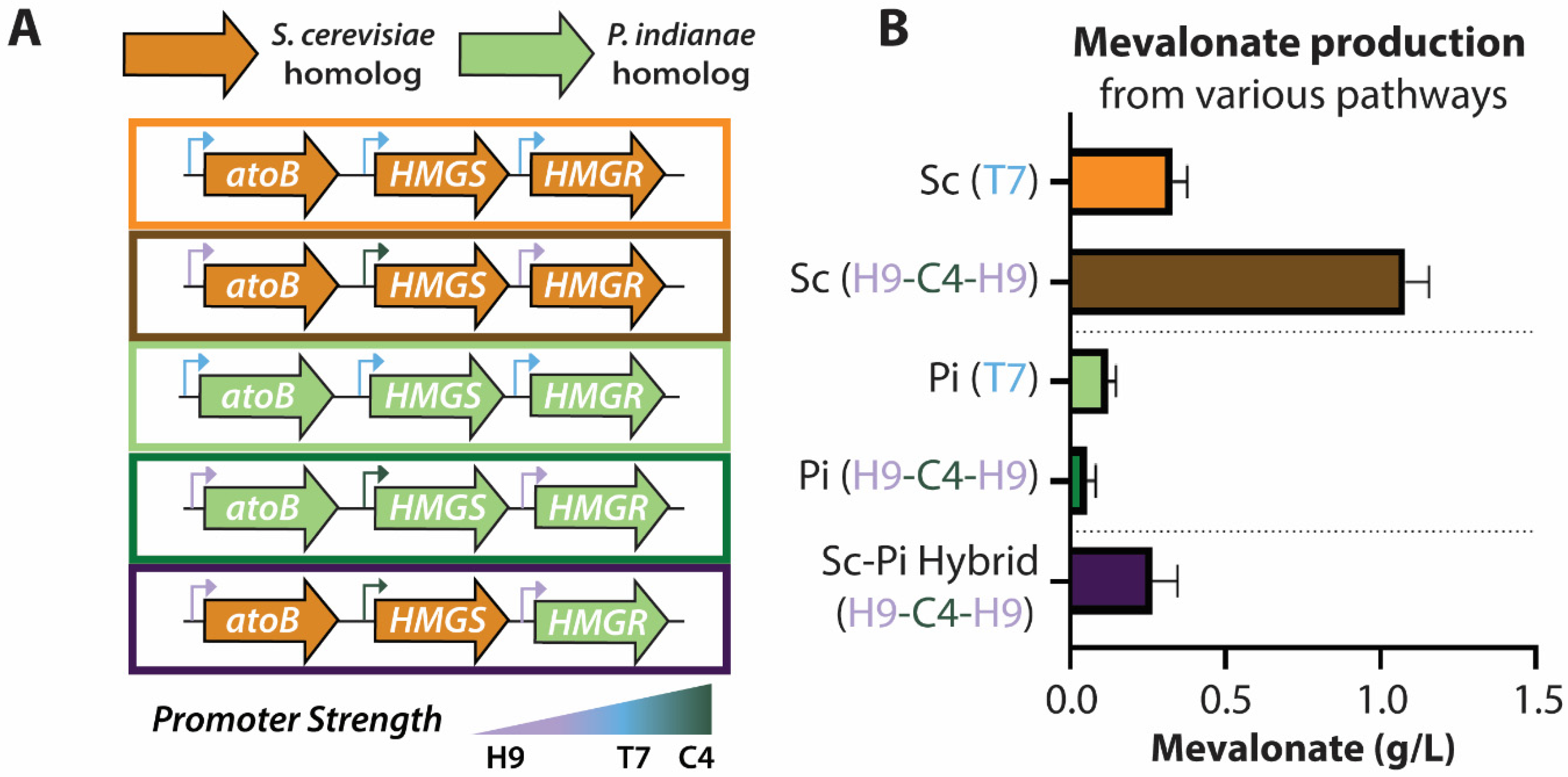
3.6. Codon Optimization Allows Heterologous Production of Mevalonate from P. indianae Enzymes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hillman, E.T.; Readnour, L.R.; Solomon, K.V. Exploiting the natural product potential of fungi with integrated-omics and synthetic biology approaches. Curr. Opin. Syst. Biol. 2017, 5, 50–56. [Google Scholar] [CrossRef]
- Flad, V.; Young, D.; Seppälä, S.; Hooker, C.; Youssef, N.; Podmirseg, S.M.; Nagler, M.; Reilly, M.; Li, Y.; Fliegerová, K.; et al. The Biotechnological Potential of Anaerobic Gut Fungi. In Genetics and Biotechnology; Benz, J.P., Schipper, K., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 413–437. ISBN 978-3-030-49924-2. [Google Scholar]
- Seppälä, S.; Wilken, S.E.; Knop, D.; Solomon, K.V.; O’Malley, M.A. The importance of sourcing enzymes from non-conventional fungi for metabolic engineering and biomass breakdown. Metab. Eng. 2017, 44, 45–59. [Google Scholar] [CrossRef]
- Hooker, C.A.; Lee, K.Z.; Solomon, K.V. Leveraging anaerobic fungi for biotechnology. Curr. Opin. Biotechnol. 2019, 59, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Henske, J.K.; Wilken, S.E.; Solomon, K.V.; Smallwood, C.R.; Shutthanandan, V.; Evans, J.E.; Theodorou, M.K.; O’Malley, M.A. Metabolic characterization of anaerobic fungi provides a path forward for bioprocessing of crude lignocellulose. Biotechnol. Bioeng. 2018, 115, 874–884. [Google Scholar] [CrossRef]
- Seppälä, S.; Yoo, J.I.; Yur, D.; O’Malley, M.A. Heterologous transporters from anaerobic fungi bolster fluoride tolerance in Saccharomyces cerevisiae. Metab. Eng. Commun. 2019, 9, e00091. [Google Scholar] [CrossRef] [PubMed]
- Perli, T.; Vos, A.M.; Bouwknegt, J.; Dekker, W.J.C.; Wiersma, S.J.; Mooiman, C.; Ortiz-Merino, R.A.; Daran, J.-M.; Pronk, J.T. Identification of Oxygen-Independent Pathways for Pyridine Nucleotide and Coenzyme A Synthesis in Anaerobic Fungi by Expression of Candidate Genes in Yeast. MBio 2021, 12, e00967-21. [Google Scholar] [CrossRef] [PubMed]
- Swift, C.; Louie, K.; Bowen, B.; Brewer, H.; Purvine, S.; Salamov, A.; Mondo, S.; Solomon, K.; Wright, A.; Northen, T.; et al. Anaerobic gut fungi are an untapped reservoir of natural products. Proc. Natl. Acad. Sci. USA 2021, 118, 1–10. [Google Scholar] [CrossRef]
- Haitjema, C.H.; Gilmore, S.P.; Henske, J.K.; Solomon, K.V.; De Groot, R.; Kuo, A.; Mondo, S.; Salamov, A.A.; LaButti, K.; Zhao, Z.; et al. A Parts List for Fungal Cellulosomes Revealed by Comparative Genomics. Nat. Microbiol. 2017, 2, 17087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, E. Special Report: The birth of biotechnology. Nature 2003, 421, 456–457. [Google Scholar] [CrossRef]
- Chi, H.; Wang, X.; Shao, Y.; Qin, Y.; Deng, Z.; Wang, L.; Chen, S. Engineering and modification of microbial chassis for systems and synthetic biology. Synth. Syst. Biotechnol. 2019, 4, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Athey, J.; Alexaki, A.; Osipova, E.; Rostovtsev, A.; Santana-Quintero, L.V.; Katneni, U.; Simonyan, V.; Kimchi-Sarfaty, C. A new and updated resource for codon usage tables. BMC Bioinform. 2017, 18, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, M.A.; Theodorou, M.K.; Kaiser, C.A. Evaluating Expression and Catalytic Activity of Anaerobic Fungal Fibroyltic Enzymes Native to Piromyces sp E2 in Saccharomyces cerevisiae. Environ. Sci. Technol. 2012, 31, 37–46. [Google Scholar] [CrossRef]
- Bernaudat, F.; Frelet-Barrand, A.; Pochon, N.; Dementin, S.; Hivin, P.; Boutigny, S.; Rioux, J.B.; Salvi, D.; Seigneurin-Berny, D.; Richaud, P.; et al. Heterologous expression of membrane proteins: Choosing the appropriate host. PLoS ONE 2011, 6, e29191. [Google Scholar] [CrossRef]
- Fernández, F.J.; Vega, M.C. Choose a Suitable Expression Host: A Survey of Available Protein Production Platforms. In Advanced Technologies for Protein Complex Production and Characterization; Vega, M.C., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 15–24. ISBN 978-3-319-27216-0. [Google Scholar]
- Heneghan, M.N.; Yakasai, A.A.; Halo, L.M.; Song, Z.; Bailey, A.M.; Simpson, T.J.; Cox, R.J.; Lazarus, C.M. First heterologous reconstruction of a complete functional fungal biosynthetic multigene cluster. ChemBioChem 2010, 11, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Saye, L.M.G.; Navaratna, T.A.; Chong, J.P.J.; O’malley, M.A.; Theodorou, M.K.; Reilly, M. The anaerobic fungi: Challenges and opportunities for industrial lignocellulosic biofuel production. Microorganisms 2021, 9, 694. [Google Scholar] [CrossRef] [PubMed]
- Wilken, S.E.; Seppälä, S.; Lankiewicz, T.S.; Saxena, M.; Henske, J.K.; Salamov, A.A.; Grigoriev, I.V.; O’Malley, M.A. Genomic and proteomic biases inform metabolic engineering strategies for anaerobic fungi. Metab. Eng. Commun. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Hooker, C.A.; Hillman, E.T.; Overton, J.C.; Ortiz-Velez, A.; Schacht, M.; Hunnicutt, A.; Mosier, N.S.; Solomon, K.V. Hydrolysis of untreated lignocellulosic feedstock is independent of S-lignin composition in newly classified anaerobic fungal isolate, Piromyces sp. UH3-1. Biotechnol. Biofuels 2018, 11, 1–14. [Google Scholar] [CrossRef]
- Hillman, E.T.; Li, M.; Hooker, C.A.; Englaender, J.A.; Wheeldon, I.; Solomon, K. V Hydrolysis of lignocellulose by anaerobic fungi produces free sugars and organic acids for two-stage fine chemical production with Kluyveromyces marxianus. Biotechnol. Prog. 2021, e3172. [Google Scholar] [CrossRef]
- Maloy, S.R. Genetic Analysis of Pathogenic Bacteria: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1996; ISBN 0879694521. [Google Scholar]
- Wood, V.; Gwilliam, R.; Rajandream, M.A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2003, 421, 871–880. [Google Scholar] [CrossRef]
- Guimaraes, J.C.; Mittal, N.; Gnann, A.; Jedlinski, D.; Riba, A.; Buczak, K.; Schmidt, A.; Zavolan, M. A rare codon-based translational program of cell proliferation. Genome Biol. 2020, 21, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.V.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014, 42, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Vansiri, A.; Bhan, N.; Koffas, M.A.G. EPathBrick: A synthetic biology platform for engineering metabolic pathways in E. coli. ACS Synth. Biol. 2012, 1, 256–266. [Google Scholar] [CrossRef]
- Martin, V.J.J.; Piteral, D.J.; Withers, S.T.; Newman, J.D.; Keasling, J.D. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat. Biotechnol. 2003, 21, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Li, S.; Jendresen, C.B.; Nielsen, A.T. Increasing production yield of tyrosine and mevalonate through inhibition of biomass formation. Process Biochem. 2016, 51, 1992–2000. [Google Scholar] [CrossRef]
- Jansen, R.; Bussemaker, H.J.; Gerstein, M. Revisiting the codon adaptation index from a whole-genome perspective: Analyzing the relationship between gene expression and codon occurrence in yeast using a variety of models. Nucleic Acids Res. 2003, 31, 2242–2251. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.D. Python Implementation of Codon Adaptation Index. J. Open Source Softw. 2018, 30, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Quax, T.E.F.; Claassens, N.J.; Söll, D.; van der Oost, J. Codon Bias as a Means to Fine-Tune Gene Expression. Mol. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Henry, I.; Sharp, P.M. Predicting gene expression level from codon usage bias. Mol. Biol. Evol. 2007, 24, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Khomtchouk, B.B.; Nonner, W. Gaussian-distributed codon frequencies of genomes. G3 Genes Genomes Genet. 2019, 9, 1449–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaney, J.L.; Steele, A.; Carmichael, R.; Rodriguez, A.; Specht, A.T.; Ngo, K.; Li, J.; Emrich, S.; Clark, P.L. Widespread position-specific conservation of synonymous rare codons within coding sequences. PLoS Comput. Biol. 2017, 13, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, P.; Brindle, J.; Stephen, J.; Plotkin, J.B.; Kudla, G. Codon usage influences fitness through RNA toxicity. Proc. Natl. Acad. Sci. USA 2018, 115, 8639–8644. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Nilsson, L.; Kurland, C.G. Co-variation of tRNA Abundance and Codon Usage inEscherichia coliat Different Growth Rates. J. Mol. Biol. 1996, 260, 649–663. [Google Scholar] [CrossRef]
- Berg, O.G.; Kurland, C.G. Growth rate-optimised tRNA abundance and codon usage. J. Mol. Biol. 1997, 270, 544–550. [Google Scholar] [CrossRef]
- Boël, G.; Letso, R.; Neely, H.; Price, W.N.; Wong, K.H.; Su, M.; Luff, J.D.; Valecha, M.; Everett, J.K.; Acton, T.B.; et al. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 2016, 529, 358–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, M.; Gong, F.; Yanofsky, C. Overexpression of tnaC of Escherichia coli inhibits growth by depleting tRNA2Pro availability. J. Bacteriol. 2006, 188, 1892–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, P.; Hemmerlin, A.; Bach, T.J.; Chye, M.L. The potential of the mevalonate pathway for enhanced isoprenoid production. Biotechnol. Adv. 2016, 34, 697–713. [Google Scholar] [CrossRef]
- Frumkin, I.; Lajoie, M.J.; Gregg, C.J.; Hornung, G.; Church, G.M.; Pilpel, Y. Codon usage of highly expressed genes affects proteome-wide translation efficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E4940–E4949. [Google Scholar] [CrossRef] [Green Version]
- McDonald, M.J.; Chou, C.H.; Swamy, K.B.S.; Huang, H.D.; Leu, J.Y. The evolutionary dynamics of tRNA-gene copy number and codon-use in E. coli. BMC Evol. Biol. 2015, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Angov, E.; Hillier, C.J.; Kincaid, R.L.; Lyon, J.A. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE 2008, 3, e2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kizer, L.; Pitera, D.J.; Pfleger, B.F.; Keasling, J.D. Application of functional genomics to pathway optimization for increased isoprenoid production. Appl. Environ. Microbiol. 2008, 74, 3229–3241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.M.; Garcia, D.E.; Redding-Johanson, A.M.; Friedland, G.D.; Chan, R.; Batth, T.S.; Haliburton, J.R.; Chivian, D.; Keasling, J.D.; Petzold, C.J.; et al. Optimization of a heterologous mevalonate pathway through the use of variant HMG-CoA reductases. Metab. Eng. 2011, 13, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; Vernacchio, V.R.; Lachance, D.M.; Lebovich, M.; Fu, L.; Shirke, A.N.; Schultz, V.L.; Cress, B.; Linhardt, R.J.; Koffas, M.A.G. EPathOptimize: A combinatorial approach for transcriptional balancing of metabolic pathways. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Morrison, J.M.; Elshahed, M.S.; Youssef, N.H. Defined enzyme cocktail from the anaerobic fungus Orpinomyces sp. Strain C1A effectively releases sugars from pretreated corn stover and switchgrass. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.A.; Toparlak, T.D.; Koffas, M.A.G. Metabolic pathway balancing and its role in the production of biofuels and chemicals. Curr. Opin. Biotechnol. 2015, 33, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.J.; Tang, X.; Moore, B.S. Genetic platforms for heterologous expression of microbial natural products. Nat. Prod. Rep. 2019, 36, 1313–1332. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Yamanaka, K.; Xu, Y.; Zhang, W.; Vlamakis, H.; Kolter, R.; Moore, B.S.; Qian, P.Y. Directed natural product biosynthesis gene cluster capture and expression in the model bacterium Bacillus subtilis. Sci. Rep. 2015, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sastalla, I.; Chim, K.; Cheung, G.Y.C.; Pomerantsev, A.P.; Leppla, S.H. Codon-optimized fluorescent proteins designed for expression in low-GC gram-positive bacteria. Appl. Environ. Microbiol. 2009, 75, 2099–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Xian, M.; Su, S.; Zhao, G.; Nie, Q.; Jiang, X.; Zheng, Y.; Liu, W. Enhancing production of bio-isoprene using hybrid MVA pathway and isoprene synthase in E. coli. PLoS ONE 2012, 7, e33509. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Niyompanich, S.; Tai, Y.-S.; Wang, J.; Bai, W.; Mahida, P.; Gao, T.; Zhang, K. Engineering of a highly efficient Escherichia coli strain for mevalonate fermentation through chromosomal integration. Appl. Environ. Microbiol. 2016, 82, 7176–7184. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hillman, E.T.; Frazier, E.M.; Shank, E.K.; Ortiz-Velez, A.N.; Englaender, J.A.; Solomon, K.V. Anaerobic Fungal Mevalonate Pathway Genomic Biases Lead to Heterologous Toxicity Underpredicted by Codon Adaptation Indices. Microorganisms 2021, 9, 1986. https://doi.org/10.3390/microorganisms9091986
Hillman ET, Frazier EM, Shank EK, Ortiz-Velez AN, Englaender JA, Solomon KV. Anaerobic Fungal Mevalonate Pathway Genomic Biases Lead to Heterologous Toxicity Underpredicted by Codon Adaptation Indices. Microorganisms. 2021; 9(9):1986. https://doi.org/10.3390/microorganisms9091986
Chicago/Turabian StyleHillman, Ethan T., Elizabeth M. Frazier, Evan K. Shank, Adrian N. Ortiz-Velez, Jacob A. Englaender, and Kevin V. Solomon. 2021. "Anaerobic Fungal Mevalonate Pathway Genomic Biases Lead to Heterologous Toxicity Underpredicted by Codon Adaptation Indices" Microorganisms 9, no. 9: 1986. https://doi.org/10.3390/microorganisms9091986
APA StyleHillman, E. T., Frazier, E. M., Shank, E. K., Ortiz-Velez, A. N., Englaender, J. A., & Solomon, K. V. (2021). Anaerobic Fungal Mevalonate Pathway Genomic Biases Lead to Heterologous Toxicity Underpredicted by Codon Adaptation Indices. Microorganisms, 9(9), 1986. https://doi.org/10.3390/microorganisms9091986