
The Effect of Common Viral Inactivation Techniques on 16S rRNA Amplicon-Based Analysis of the Gut Microbiota


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Viral Inactivation Simulation and DNA Extraction
2.2.1. Control Extractions
2.2.2. SDS
2.2.3. Holder Pasteurization
2.2.4. TRIzol
2.2.5. Buffer AVL
2.2.6. Buffer AVL with Pasteurization
2.2.7. 16S rRNA Amplification and Sequencing
2.3. Informatics
2.4. Statistical Analysis
3. Results
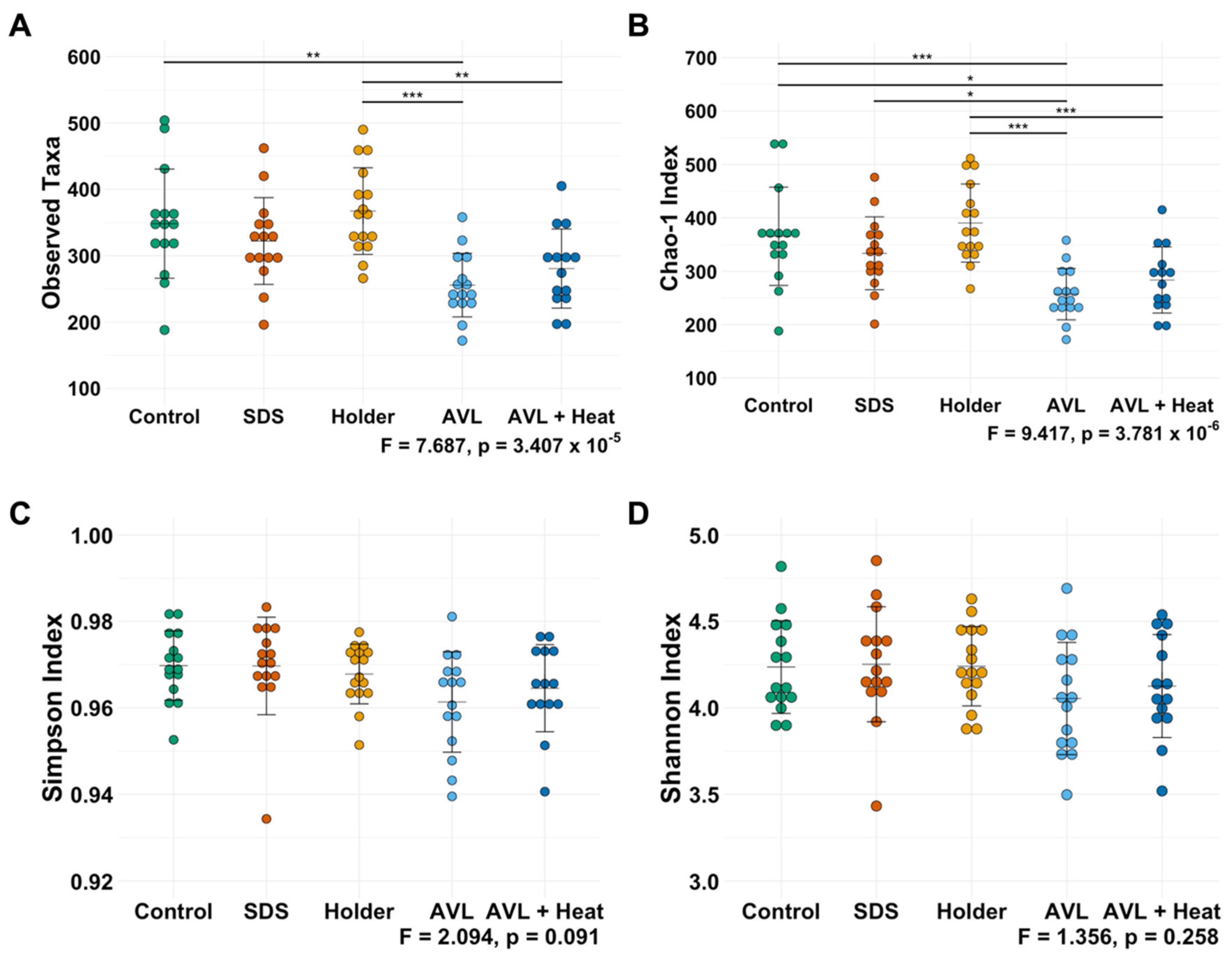
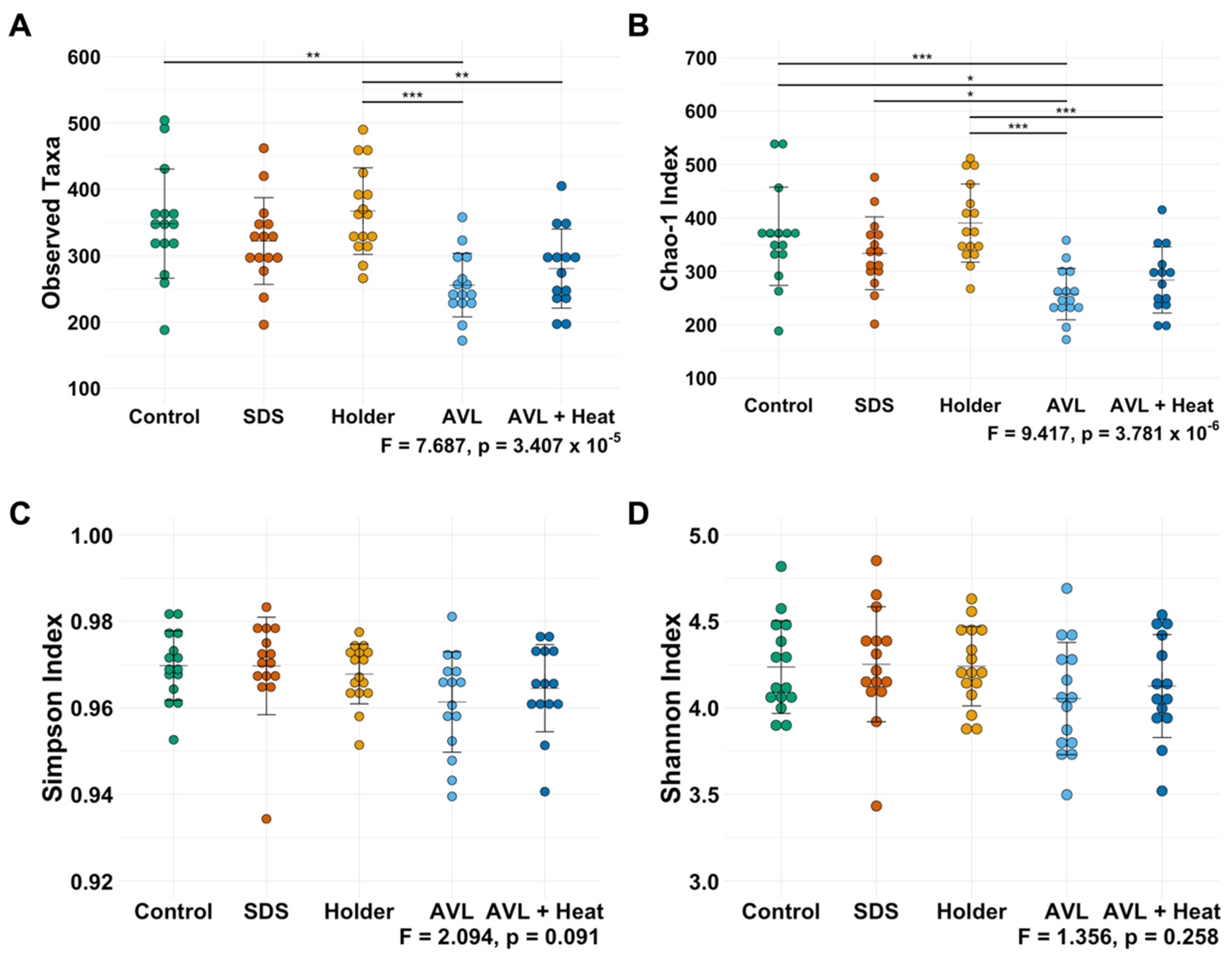
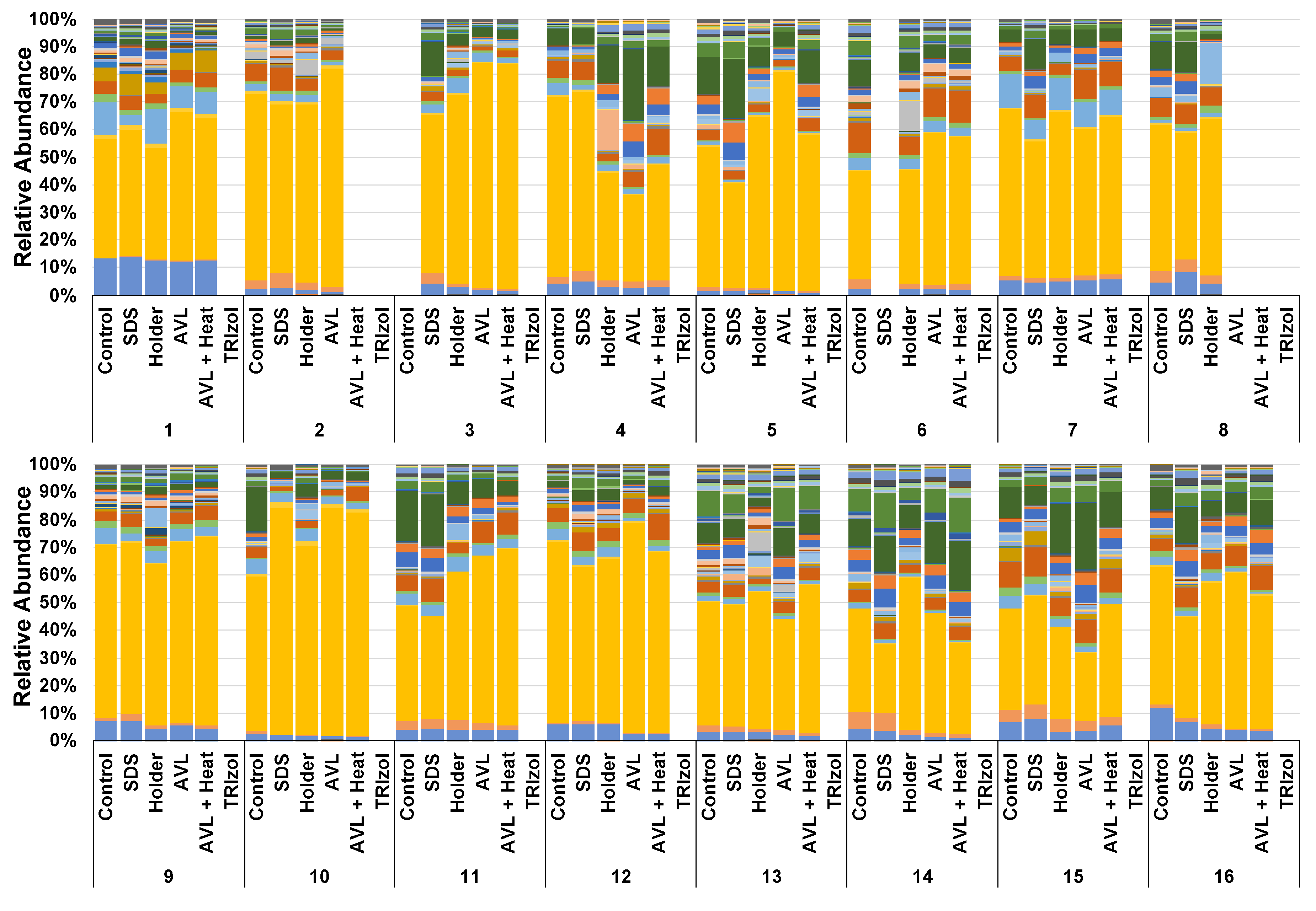
3.1. Not All Viral Inactivation Methods Yield Sequencing-Quality DNA
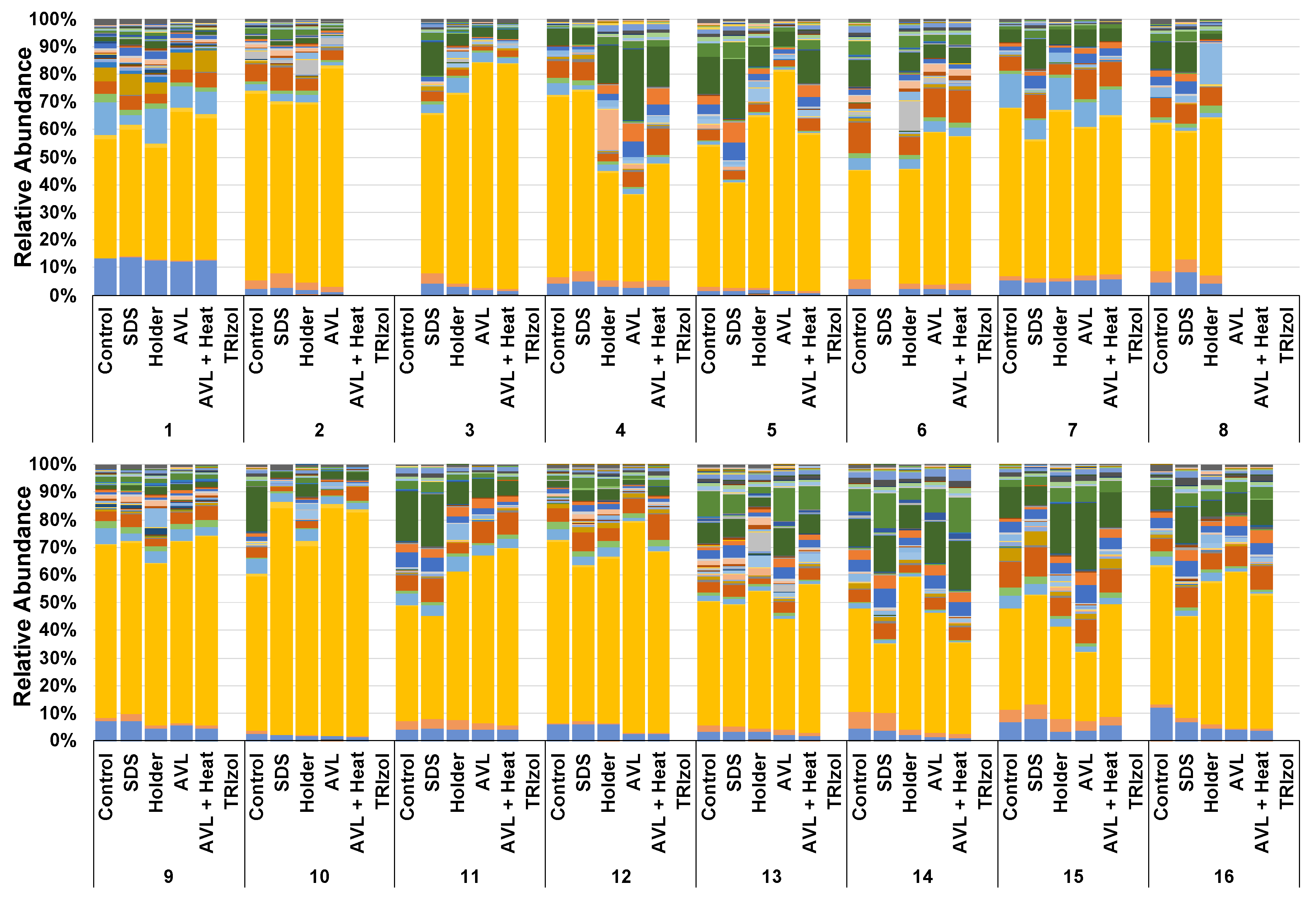
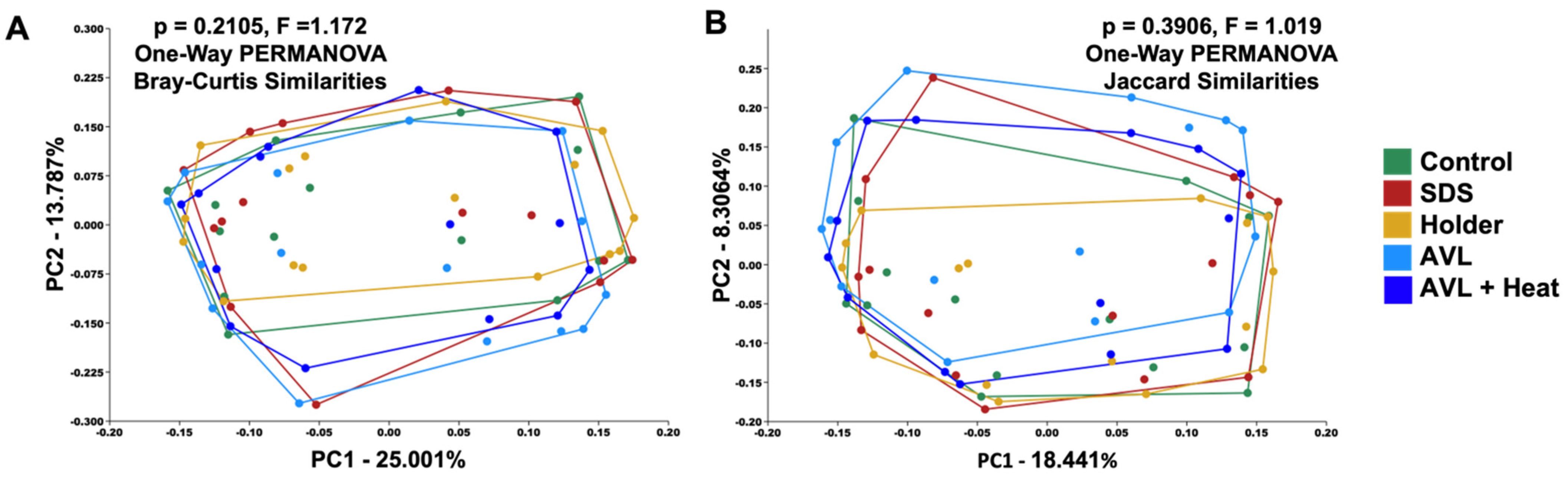
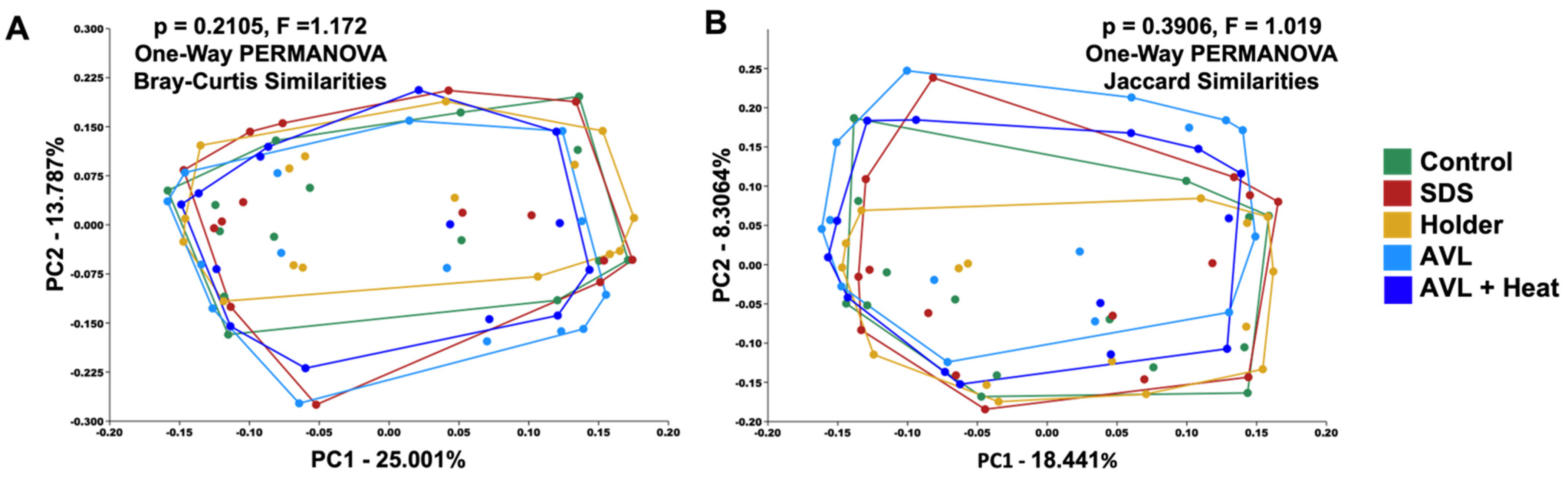
3.2. Treatment-Dependent Effects on Microbial Richness but Not Distribution or Beta Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, D.; Leung, R.K.-K.; Guan, W.; Au, W.W. Involvement of Gut Microbiome in Human Health and Disease: Brief Overview, Knowledge Gaps and Research Opportunities. Gut Pathog. 2018, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Ma, W.-T.; Pang, M.; Fan, Q.-L.; Hua, J.-L. The Commensal Microbiota and Viral Infection: A Comprehensive Review. Front. Immunol. 2019, 10, 1551. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, J.; Guo, L.; Yao, H.; Wang, L.; Xia, X.; Zhang, W. Fecal Viral Shedding in COVID-19 Patients: Clinical Significance, Viral Load Dynamics and Survival Analysis. Virus Res. 2020, 289, 198147. [Google Scholar] [CrossRef] [PubMed]
- Koçer, Z.A.; Obenauer, J.; Zaraket, H.; Zhang, J.; Rehg, J.E.; Russell, C.J.; Webster, R.G. Fecal Influenza in Mammals: Selection of Novel Variants. J. Virol. 2013, 87, 11476–11486. [Google Scholar] [CrossRef] [Green Version]
- Park, S.L.; Huang, Y.-J.S.; Hsu, W.-W.; Hettenbach, S.M.; Higgs, S.; Vanlandingham, D.L. Virus-Specific Thermostability and Heat Inactivation Profiles of Alphaviruses. J. Virol. Methods 2016, 234, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Hamprecht, K.; Maschmann, J.; Müller, D.; Dietz, K.; Besenthal, I.; Goelz, R.; Middeldorp, J.M.; Speer, C.P.; Jahn, G. Cytomegalovirus (CMV) Inactivation in Breast Milk: Reassessment of Pasteurization and Freeze-Thawing. Pediatr. Res. 2004, 56, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batéjat, C.; Grassin, Q.; Manuguerra, J.-C.; Leclercq, I. Heat Inactivation of the Severe Acute Respiratory Syndrome Coronavirus 2. J. Biosaf. Biosecur. 2021, 3, 1–3. [Google Scholar] [CrossRef]
- García-González, I.; Corona-Cervantes, K.; Hernández-Quiroz, F.; Villalobos-Flores, L.E.; Galván-Rodríguez, F.; Romano, M.C.; Miranda-Brito, C.; Piña-Escobedo, A.; Borquez-Arreortúa, F.G.; Rangel-Calvillo, M.N.; et al. The Effect of Holder Pasteurization on the Diversity of the Human Milk Bacterial Microbiota Using High-Throughput DNA Sequencing. J. Hum. Lact. 2021, 089033442110119. [Google Scholar] [CrossRef]
- Urdaneta, S.; Wigdahl, B.; Neely, E.B.; Berlin, C.M.; Schengrund, C.-L.; Lin, H.-M.; Howett, M.K. Inactivation of HIV-1 in Breast Milk by Treatment with the Alkyl Sulfate Microbicide Sodium Dodecyl Sulfate (SDS). Retrovirology 2005, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Patterson, E.I.; Prince, T.; Anderson, E.R.; Casas-Sanchez, A.; Smith, S.L.; Cansado-Utrilla, C.; Solomon, T.; Griffiths, M.J.; Acosta-Serrano, Á.; Turtle, L.; et al. Methods of Inactivation of SARS-CoV-2 for Downstream Biological Assays. J. Infect. Dis. 2020, 222, jiaa507. [Google Scholar] [CrossRef]
- Ericsson, A.C.; Davis, J.W.; Spollen, W.; Bivens, N.; Givan, S.; Hagan, C.E.; McIntosh, M.; Franklin, C.L. Effects of Vendor and Genetic Background on the Composition of the Fecal Microbiota of Inbred Mice. PLoS ONE 2015, 10, e0116704. [Google Scholar] [CrossRef] [Green Version]
- Blow, J.A.; Dohm, D.J.; Negley, D.L.; Mores, C.N. Virus Inactivation by Nucleic Acid Extraction Reagents. J. Virol. Methods 2004, 119, 195–198. [Google Scholar] [CrossRef]
- Smither, S.J.; Weller, S.A.; Phelps, A.; Eastaugh, L.; Ngugi, S.; O’Brien, L.M.; Steward, J.; Lonsdale, S.G.; Lever, M.S. Buffer AVL Alone Does Not Inactivate Ebola Virus in a Representative Clinical Sample Type. J. Clin. Microbiol. 2015, 53, 3148–3154. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.L.; Ericsson, A.C.; Lloyd, K.C.K.; Grimsrud, K.N.; Rogala, A.R.; Godfrey, V.L.; Nielsen, J.N.; Franklin, C.L. Development of Outbred CD1 Mouse Colonies with Distinct Standardized Gut Microbiota Profiles for Use in Complex Microbiota Targeted Studies. Sci. Rep. 2018, 8, 10107. [Google Scholar] [CrossRef]
- Pitino, M.A.; O’Connor, D.L.; McGeer, A.J.; Unger, S. The Impact of Thermal Pasteurization on Viral Load and Detectable Live Viruses in Human Milk and Other Matrices: A Rapid Review. Appl. Physiol. Nutr. Metab. 2020, 99, 1–17. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global Patterns of 16S RRNA Diversity at a Depth of Millions of Sequences per Sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and Microbial Differential Abundance Strategies Depend upon Data Characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) Format or: How I Learned to Stop Worrying and Love the Ome-Ome. Gigascience 2012, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaehler, B.D.; Bokulich, N.A.; McDonald, D.; Knight, R.; Caporaso, J.G.; Huttley, G.A. Species Abundance Information Improves Sequence Taxonomy Classification Accuracy. Nat. Commun. 2019, 10, 4643. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible Sequence Taxonomy Reference Database Management for the Masses. Biorxiv 2020. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- RC Team. R: A Language and Environment for Statistical Computing; RC Team: Vienna, Austria, 2019. [Google Scholar]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, gkab382. [Google Scholar] [CrossRef]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A Tool for Visualizing High-Throughput Microbial Community Data. Gigascience 2013, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Ngo, K.A.; Jones, S.A.; Church, T.M.; Fuschino, M.E.; George, K.S.; Lamson, D.M.; Maffei, J.; Kramer, L.D.; Ciota, A.T. Unreliable Inactivation of Viruses by Commonly Used Lysis Buffers. Appl. Biosaf. 2017, 22, 56–59. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Inactivation Method | DNA Yield (ng DNA/mg Feces, n = 16) | Successful 16S Sequencing (≥10,000 Reads) | Features per Sample Post-Denoising (Mean ± SD, n = 16) | Sample Number Passing 40,000 Feature Rarefication Filter |
|---|---|---|---|---|
| Control | 135.9 ± 106.2 | 15/16 | 99,572 ± 34,364 | 15/16 |
| SDS | 242.0 ± 186.0 | 16/16 | 84,754 ± 17,681 | 15/16 |
| TRIzol | 18.7 ± 19.3 | 0/16 | 0 | 0/16 |
| Holder | 65.3 ± 29.5 | 16/16 | 111,909 ± 17,831 | 16/16 |
| AVL | 0.2 ± 0.5 | 16/16 | 62,508 ± 18,162 | 15/16 |
| AVL + Heat | 0.4 ± 0.8 | 16/16 | 72,796 ± 21,625 | 14/16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McAdams, Z.; Gustafson, K.; Ericsson, A. The Effect of Common Viral Inactivation Techniques on 16S rRNA Amplicon-Based Analysis of the Gut Microbiota. Microorganisms 2021, 9, 1755. https://doi.org/10.3390/microorganisms9081755
McAdams Z, Gustafson K, Ericsson A. The Effect of Common Viral Inactivation Techniques on 16S rRNA Amplicon-Based Analysis of the Gut Microbiota. Microorganisms. 2021; 9(8):1755. https://doi.org/10.3390/microorganisms9081755
Chicago/Turabian StyleMcAdams, Zachary, Kevin Gustafson, and Aaron Ericsson. 2021. "The Effect of Common Viral Inactivation Techniques on 16S rRNA Amplicon-Based Analysis of the Gut Microbiota" Microorganisms 9, no. 8: 1755. https://doi.org/10.3390/microorganisms9081755
APA StyleMcAdams, Z., Gustafson, K., & Ericsson, A. (2021). The Effect of Common Viral Inactivation Techniques on 16S rRNA Amplicon-Based Analysis of the Gut Microbiota. Microorganisms, 9(8), 1755. https://doi.org/10.3390/microorganisms9081755