
Comparative Genomics Analyses Reveal the Differences between B. longum subsp. infantis and B. longum subsp. longum in Carbohydrate Utilisation, CRISPR-Cas Systems and Bacteriocin Operons

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. B. Longum Strain, Genonic Sequencing and Data Assembly
2.2. Average Nucleotide Identity (ANI) Values
2.3. Phylogenetic Analyses
2.4. Pan-Genome and Core-Genome Analysis
2.5. Whole Genome and Orthologous Gene Comparison
2.6. Genotype and Phenotype Analysis of Carbohydrate Metabolism
2.7. CRISPR-Cas Systems Prediction
2.8. Bacteriocin Prediction
2.9. Statistical Analysis
3. Results
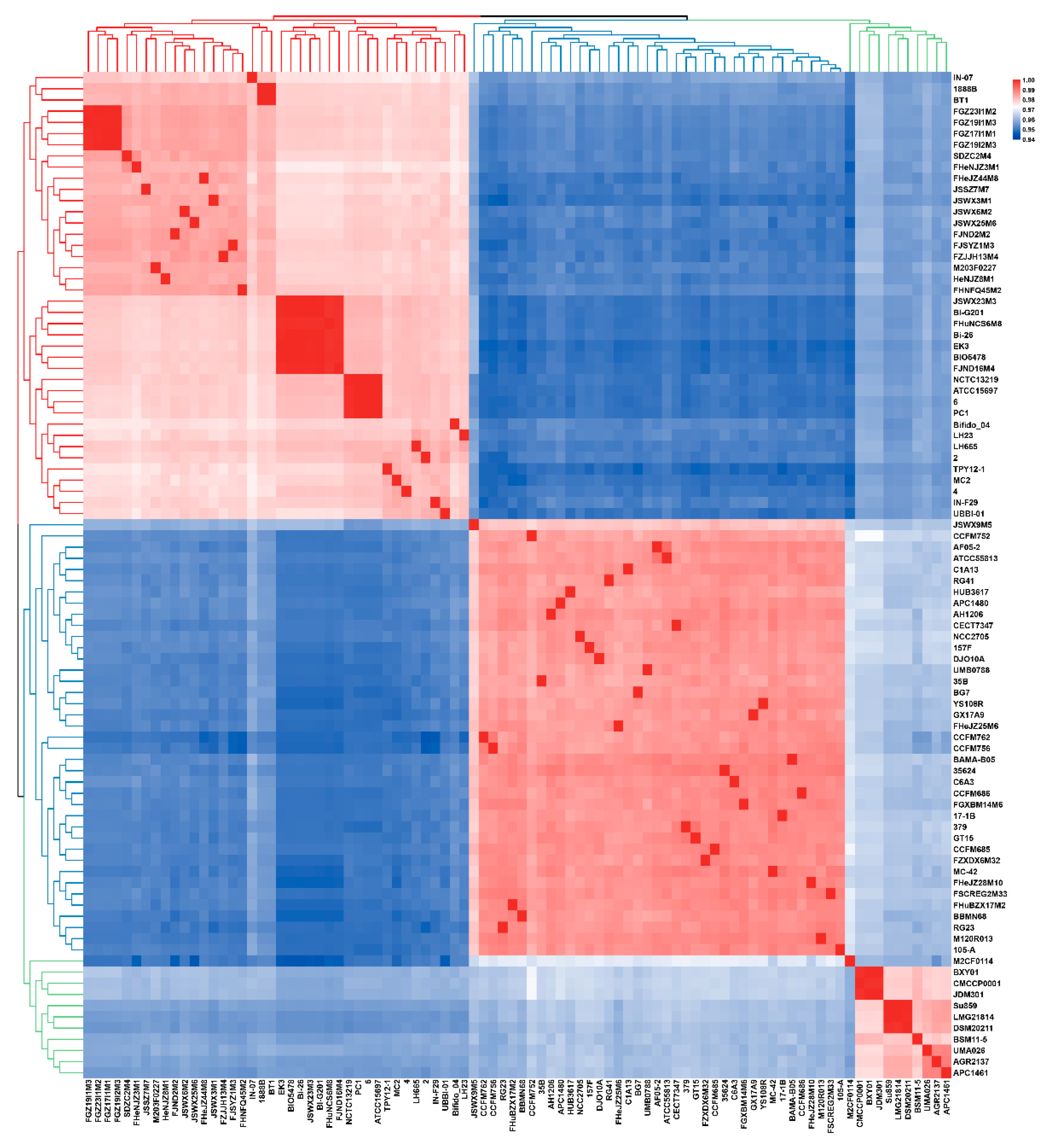
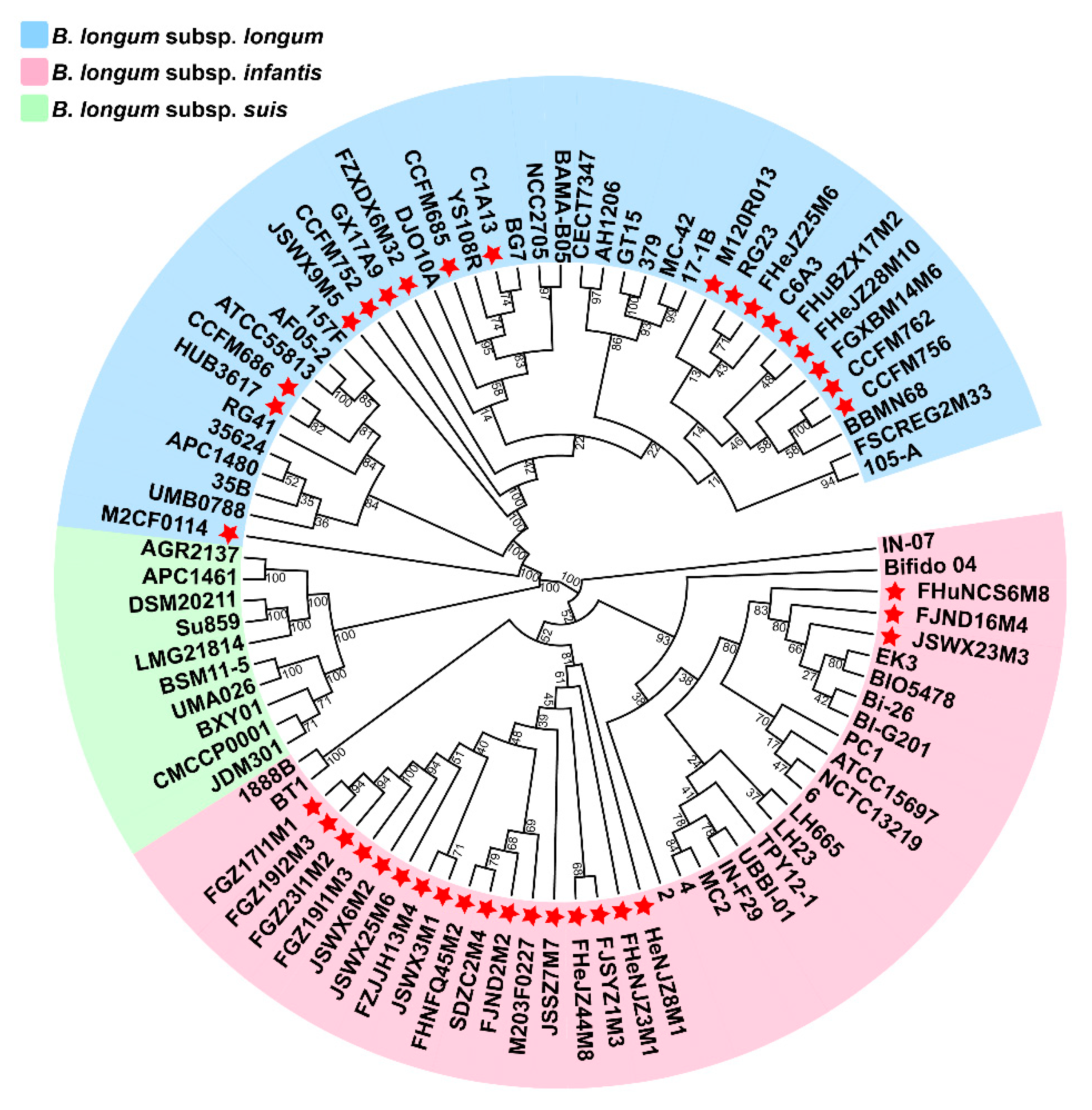
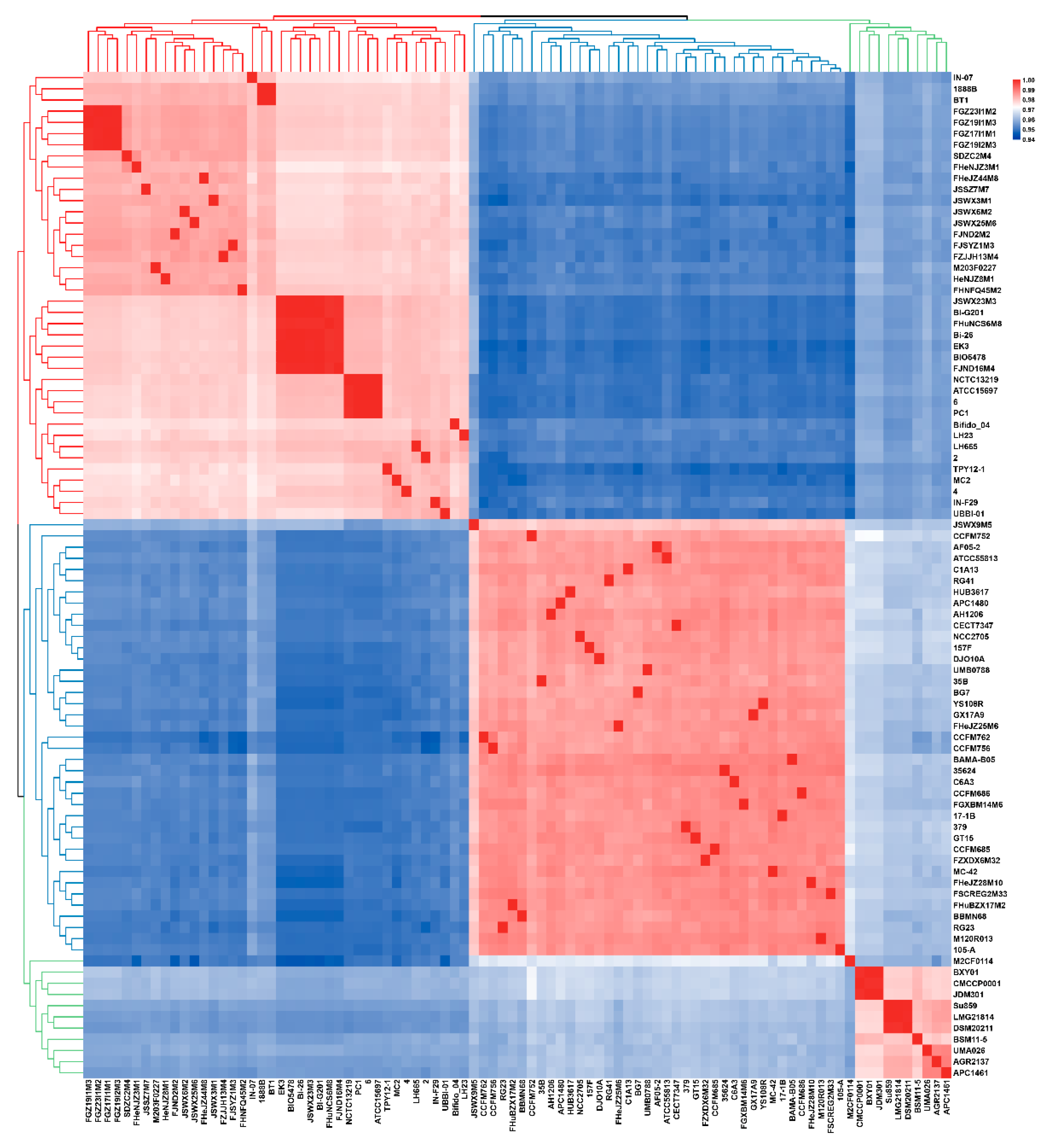
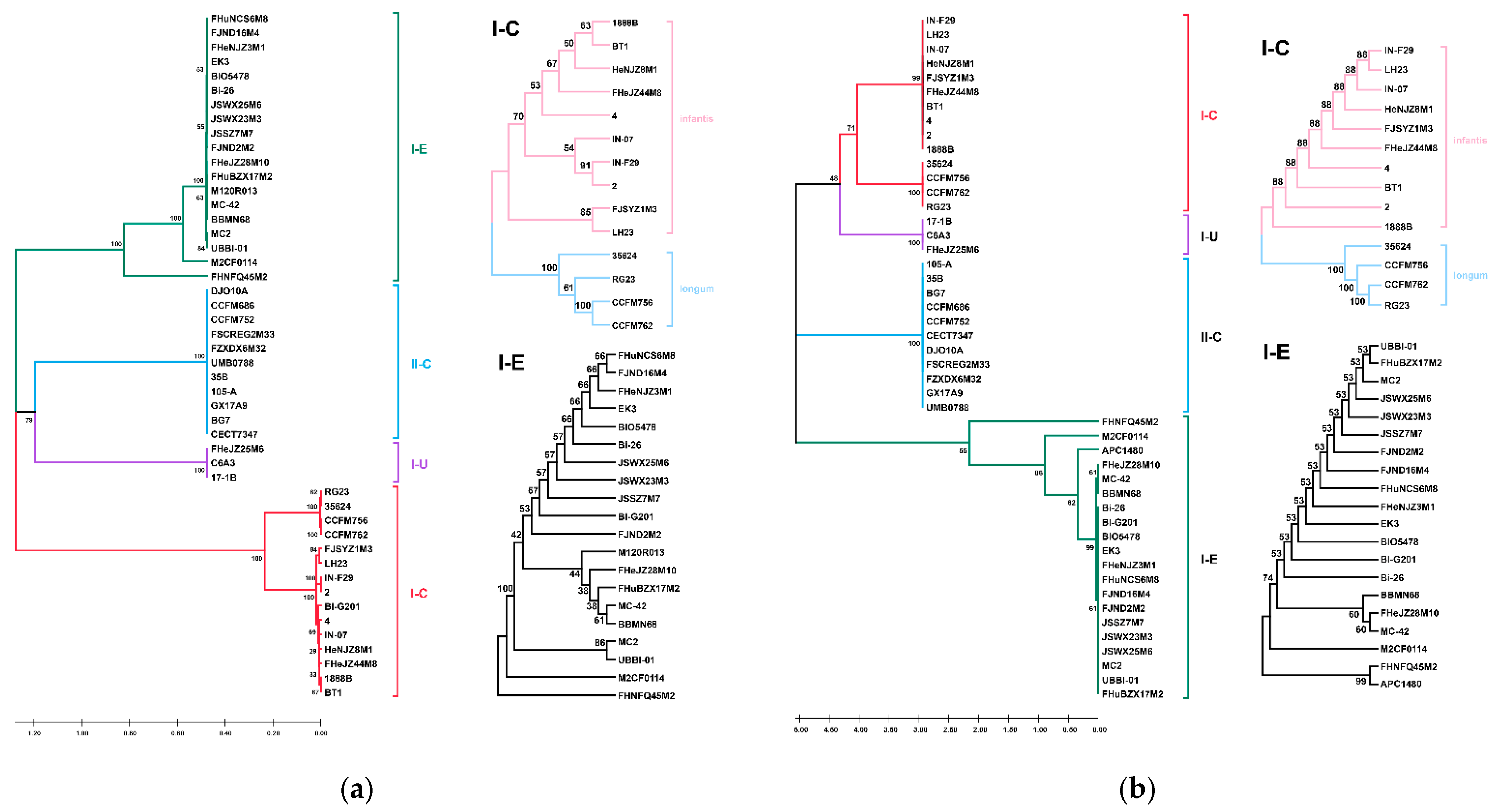
3.1. ANI and Phylogenetic Analysis of B. longum
3.2. General Genome Features of B. longum subsp. infantis and B. longum subsp. longum
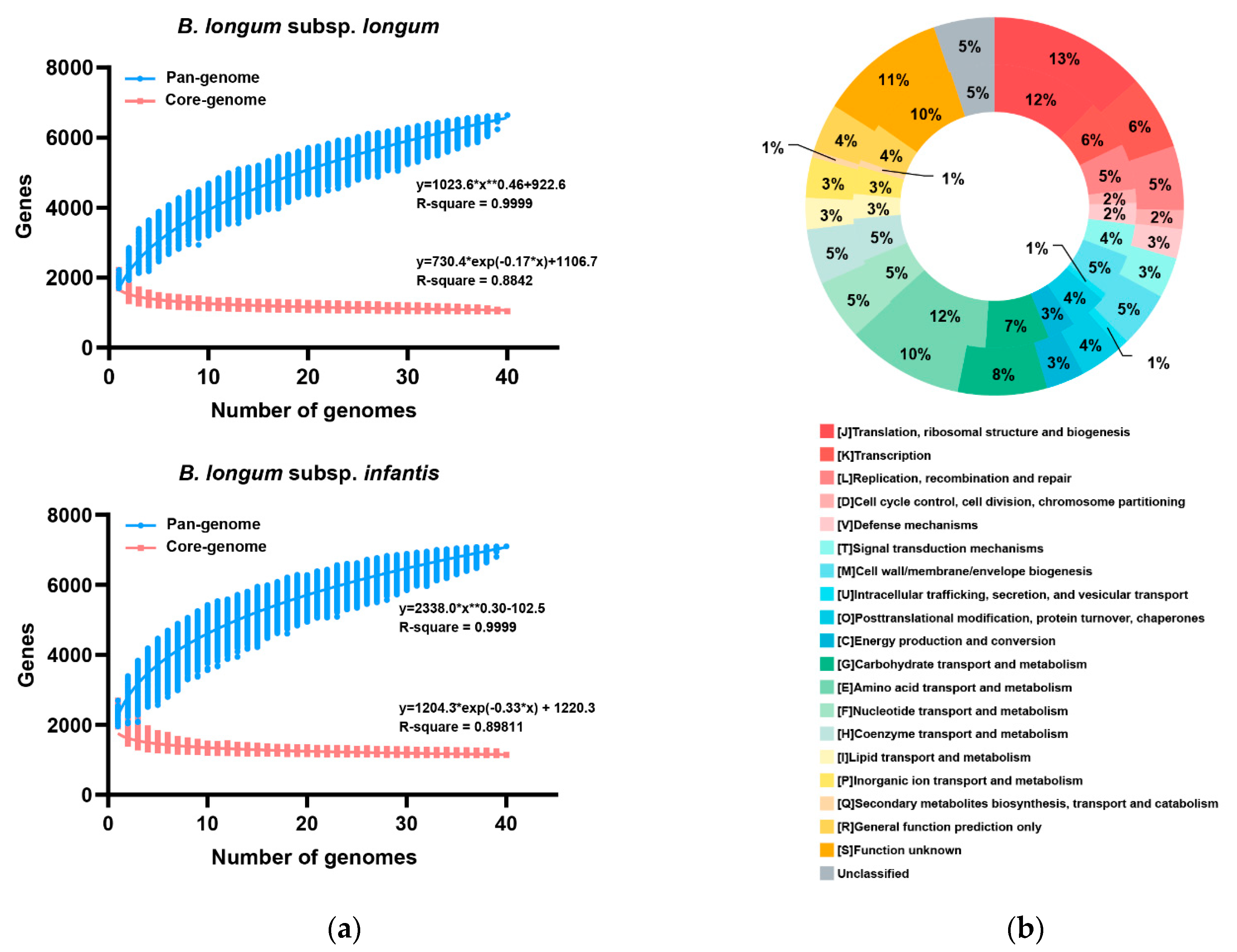
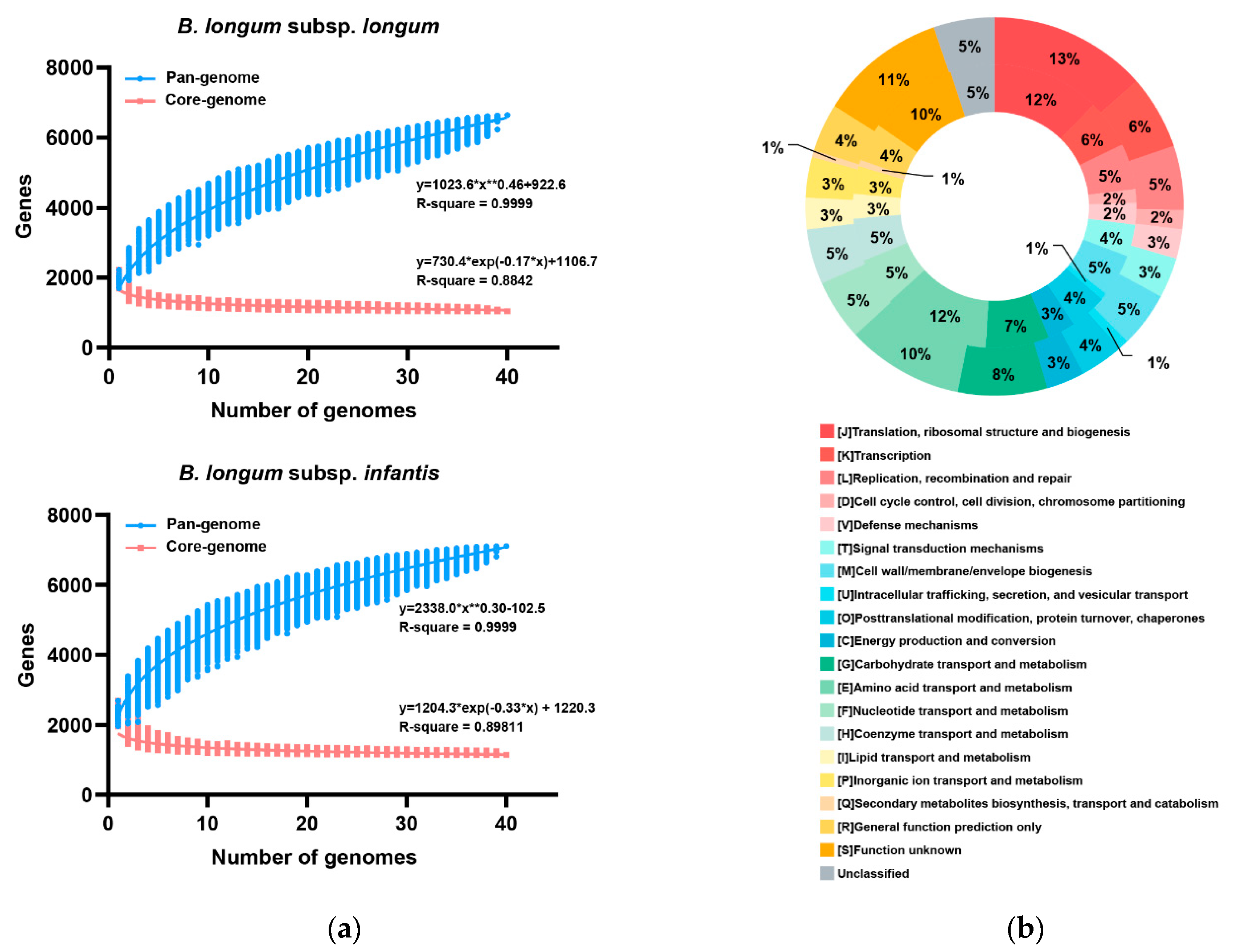
3.3. Pan- and Core-Genome of B. longum subsp. infantis and B. longum subsp. longum
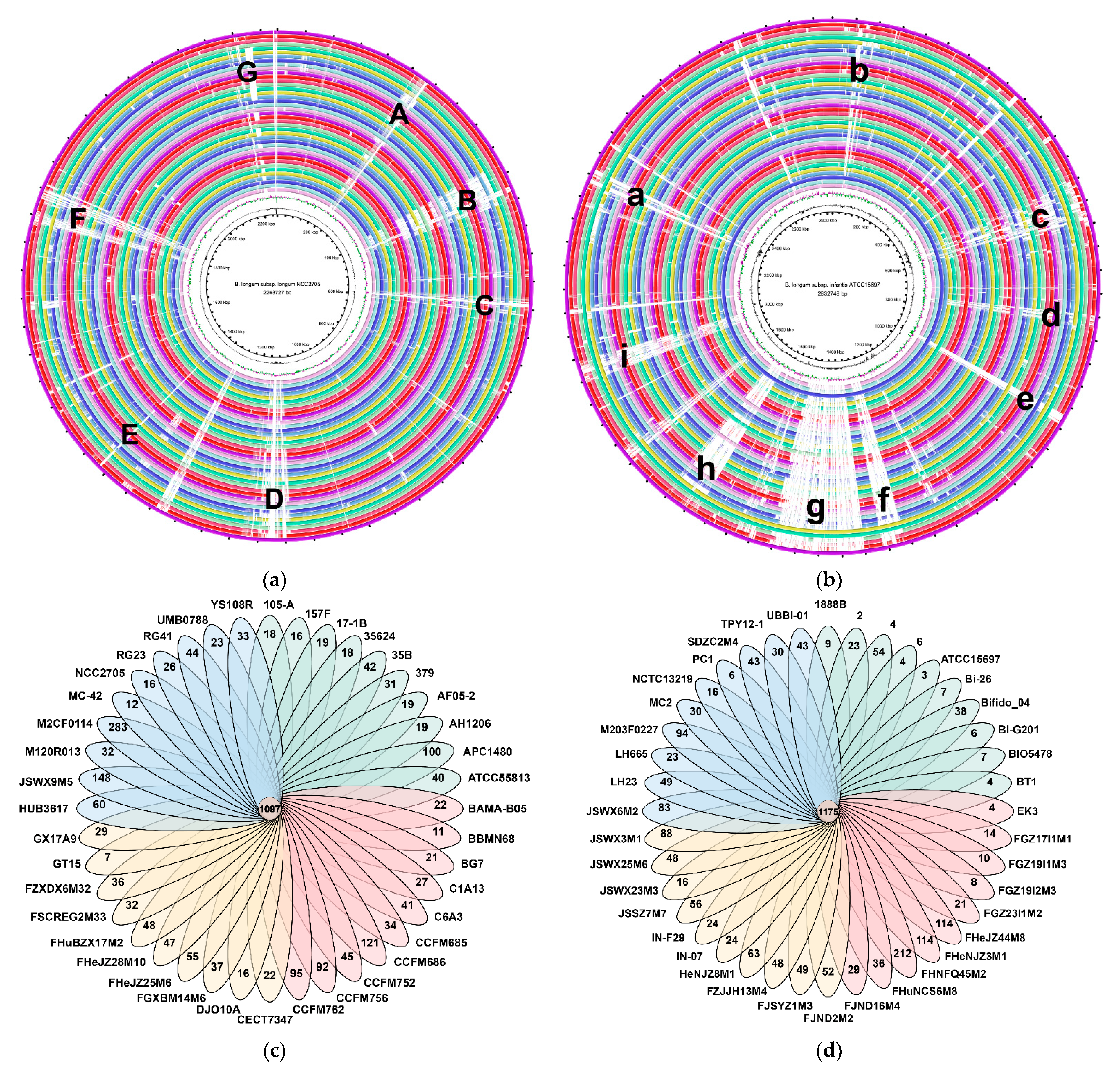
3.4. Whole Genome and Orthologous Gene Comparison
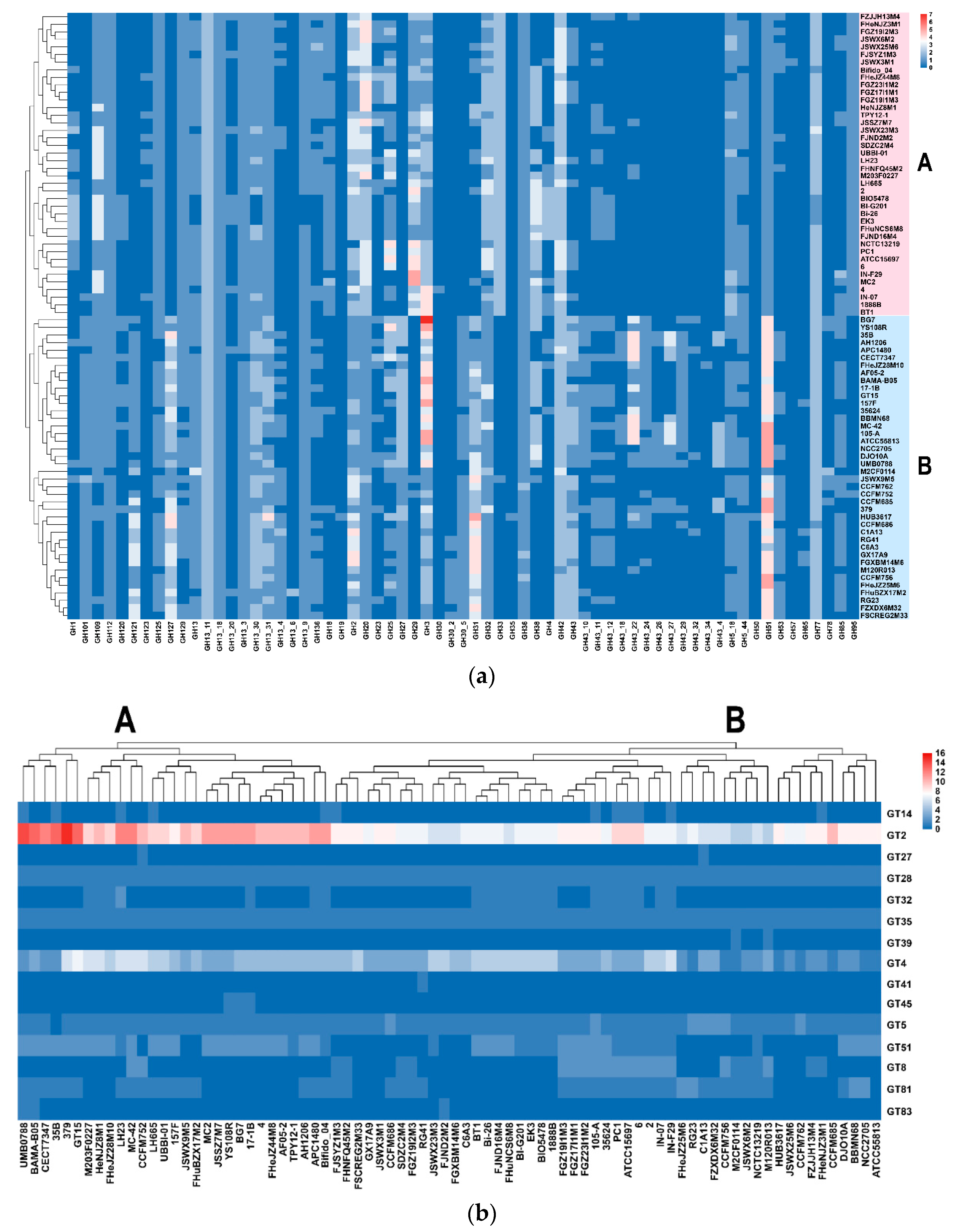
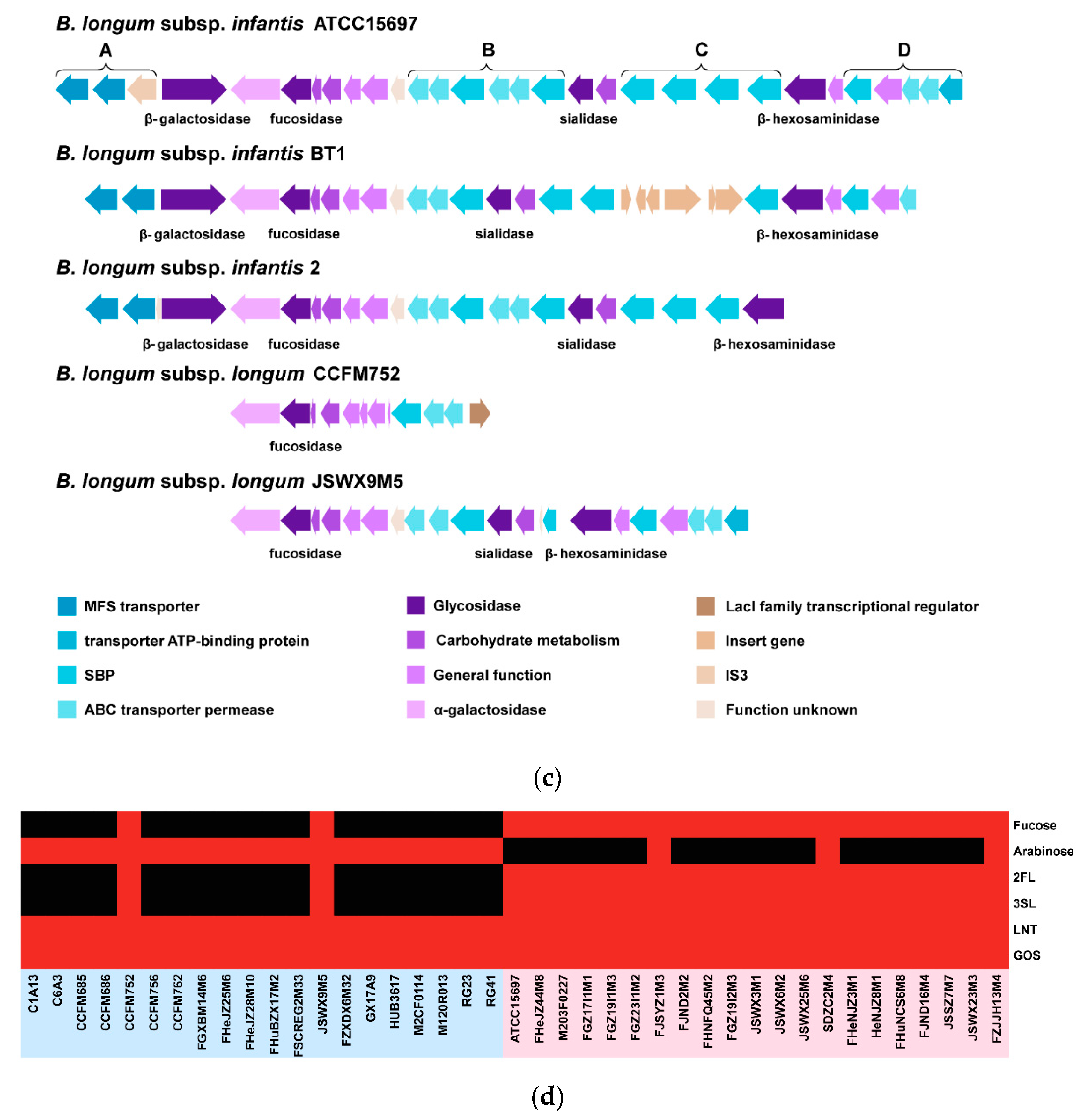
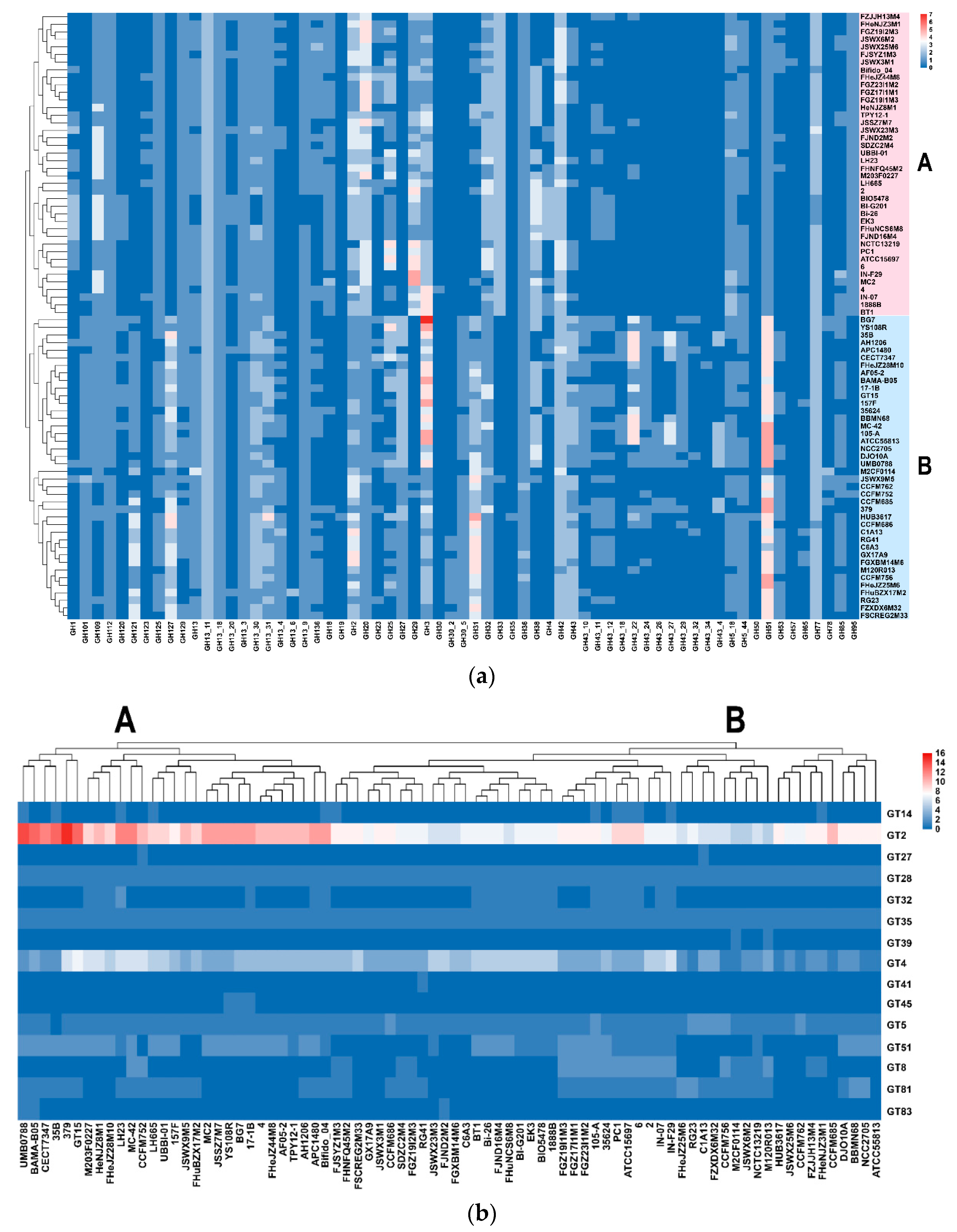
3.5. Carbohydrate Utilization Genotype and Phenotype of B. longum subsp. infantis and B. longum subsp. longum
3.6. Predicted EPS Gene Clusters in B. longum subsp. infantis and B. longum subsp. longum
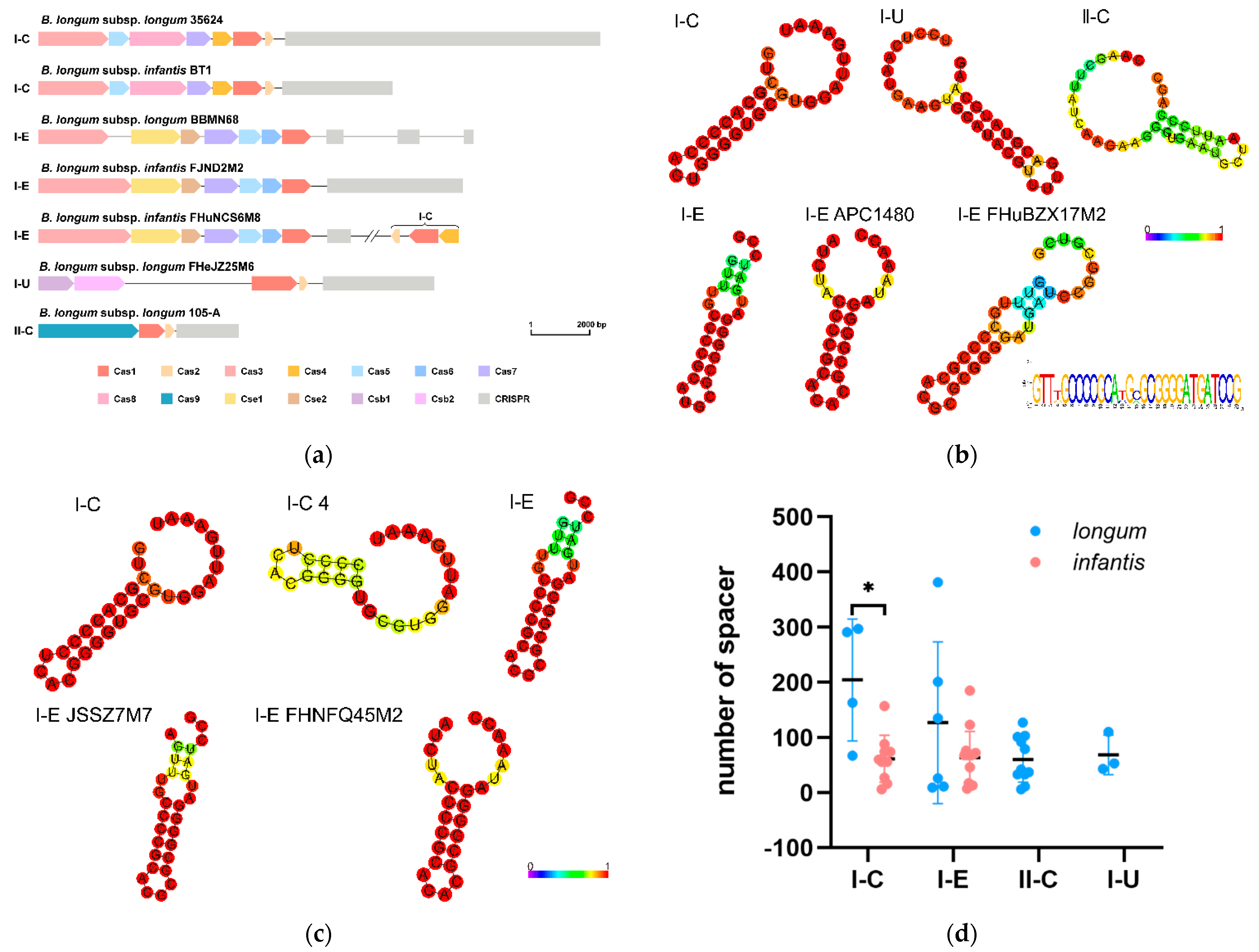
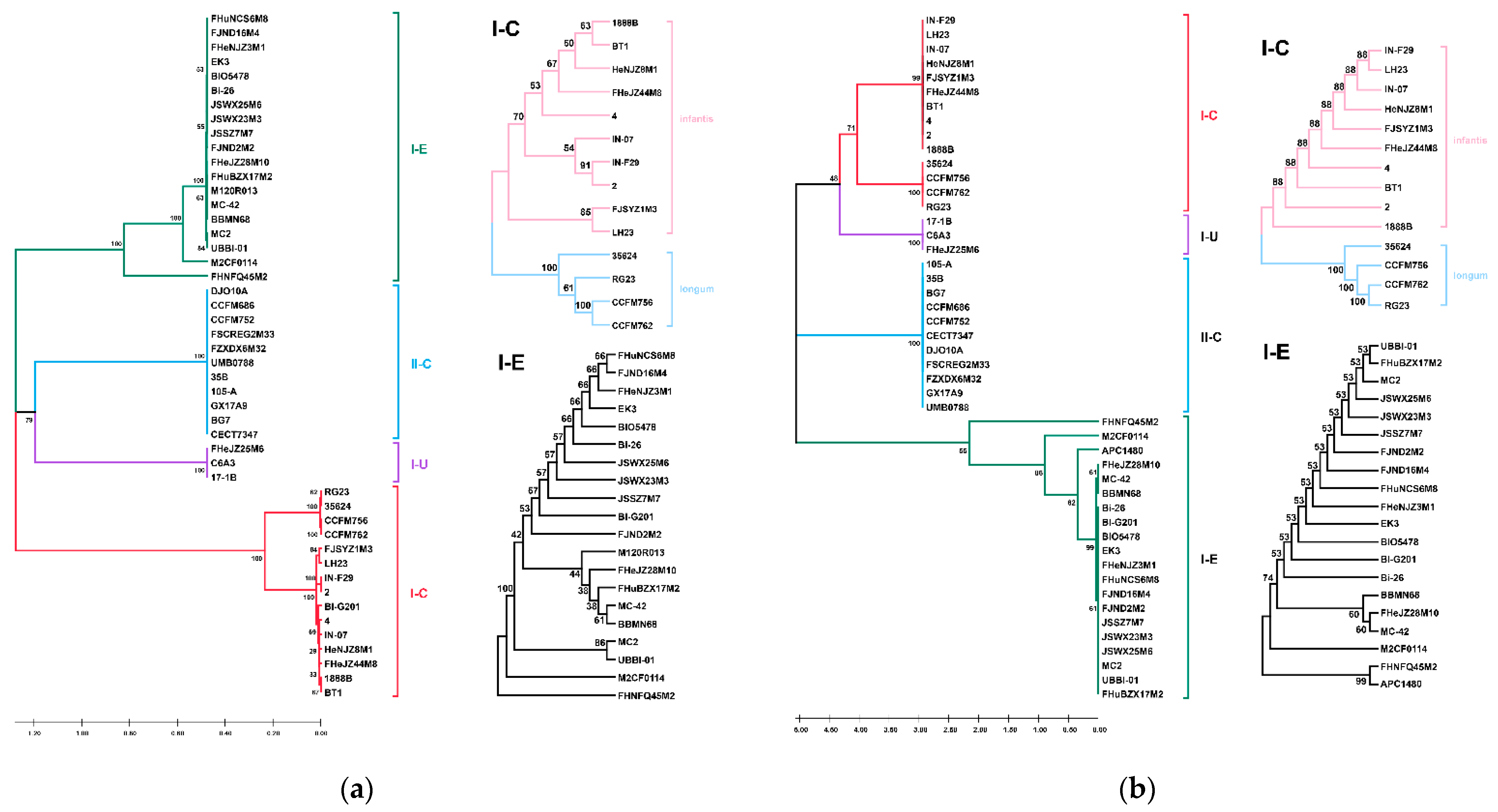
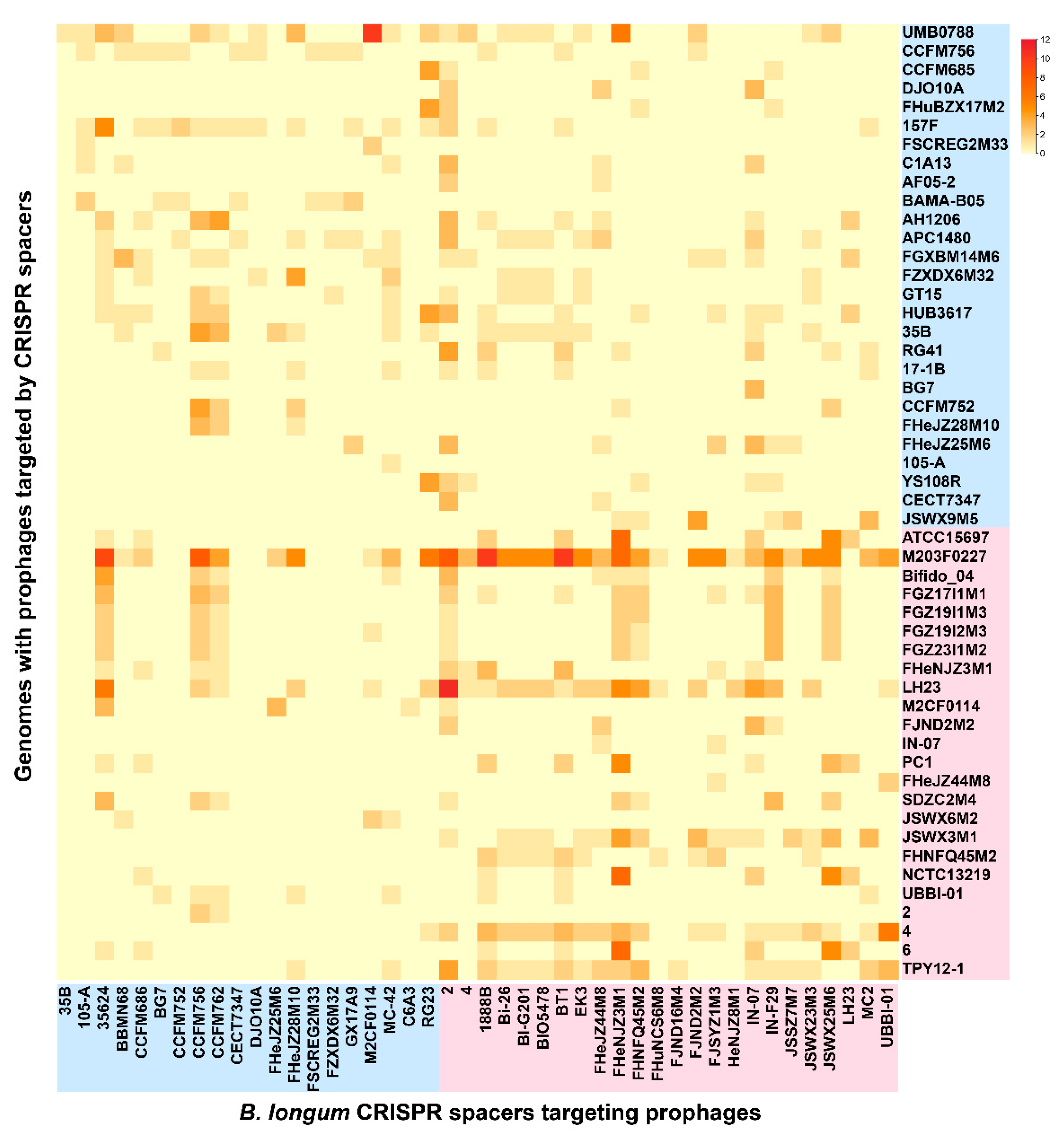
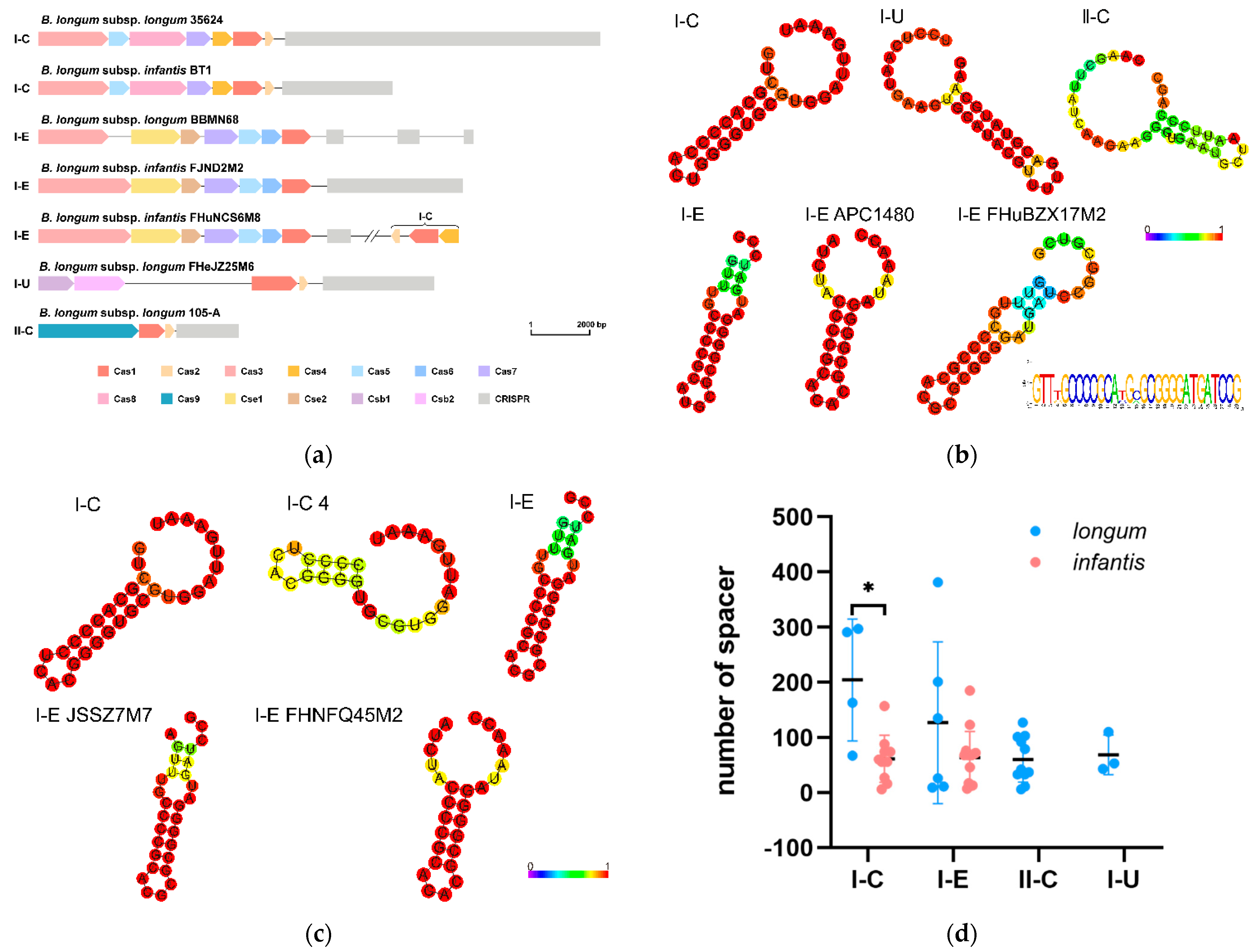
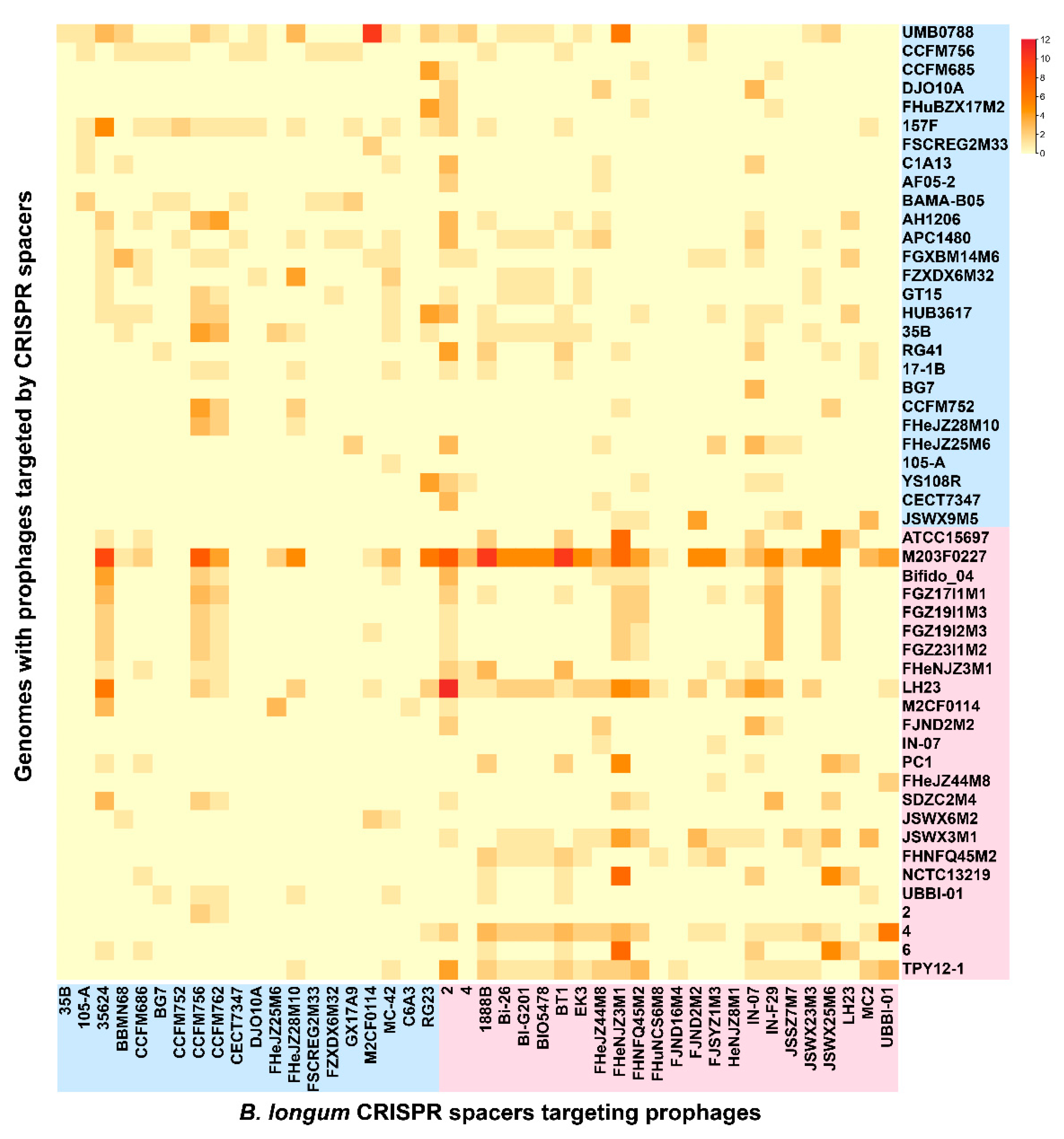
3.7. CRISPR-Cas Systems in B. longum subsp. infantis and B. longum subsp. longum
3.8. Bacteriocin Operons in B. longum subsp. infantis and B. longum subsp. longum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattarelli, P.; Bonaparte, C.; Pot, B.; Biavati, B. Proposal to reclassify the three biotypes of Bifidobacterium longum as three subspecies: Bifidobacterium longum subsp. longum subsp. nov., Bifidobacterium longum subsp. infantis comb. nov. and Bifidobacterium longum subsp. suis comb. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 767–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matteuzzi, D.; Crociani, F.; Zani, O.; Trovatelli, L.D. Bifidobacterium suis n. sp.: A new species of the genus Bifidobacterium isolated from pig faces. J. Basic Microbiol. 1971, 11, 387–395. [Google Scholar] [CrossRef]
- Groeger, D.; O’Mahony, L.; Murphy, E.F.; Bourke, J.F.; Dinan, T.; Kiely, B.; Shanahan, F.; Quigley, E.M. Bifidobacterium infantis 35624 modulates host inflammatory processes beyond the gut. Gut Microbes 2013, 4, 325–339. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, E.; Ogita, T.; Miyamoto, J.; Kawamoto, S.; Morita, H.; Ohno, H.; Suzuki, T.; Tanabe, S. Bifidobacterium longum alleviates dextran sulfate sodium-induced colitis by suppressing il-17a response: Involvement of intestinal epithelial costimulatory molecules. PLoS ONE 2013, 8, e79735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barba-Vidal, E.; Castillejos, L.; Colom, P.L.; Urgell, M.R.; Muñoz, J.A.M.; Martín-Orúe, S.M. Evaluation of the probiotic strain Bifidobacterium longum subsp. Infantis CECT 7210 capacities to improve health status and fight digestive pathogens in a piglet model. Front. Microbiol. 2017, 8, 533. [Google Scholar] [CrossRef] [Green Version]
- Sela, D.A.; Chapman, J.; Adeuya, A.; Kim, J.; Chen, F.; Whitehead, T.R.; Lapidus, A.; Rokhsar, D.; Lebrilla, C.B.; German, J.B.; et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 18964–18969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakata, S. Unification of Bifidobacterium infantis and Bifidobacterium suis as Bifidobacterium longum. Int. J. Syst. Evol. Microbiol. 2002, 52, 1945–1951. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, A.V.; Efimov, B.A.; Smeianov, V.; Kafarskaia, L.I.; Pikina, A.P.; Shkoporov, A. Intraspecies genomic diversity and long-term persistence of Bifidobacterium longum. PLoS ONE 2015, 10, e0135658. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, A.; Bottacini, F.; Motherway, M.O.; Van Sinderen, D. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction-modification systems. BMC Genom. 2015, 16, 832. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Cantabrana, C.; Crawley, A.; Sanchez, B.; Barrangou, R. Characterization and exploitation of CRISPR loci in Bifidobacterium longum. Front. Microbiol. 2017, 8, 1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arboleya, S.; Bottacini, F.; O’Connell-Motherway, M.; Ryan, C.A.; Ross, R.P.; Van Sinderen, D.; Stanton, C. Gene-trait matching across the Bifidobacterium longum pan-genome reveals considerable diversity in carbohydrate catabolism among human infant strains. BMC Genom. 2018, 19, 33. [Google Scholar] [CrossRef]
- Jiang, J.; Yang, B.; Ross, R.; Stanton, C.; Zhao, J.; Zhang, H.; Chen, W. Comparative genomics of pediococcus pentosaceus isolated from different niches reveals genetic diversity in carbohydrate metabolism and immune system. Front. Microbiol. 2020, 11, 253. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Chen, F. OrthoMCL-DB: Querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006, 34, D363–D368. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.-H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef] [Green Version]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [Green Version]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.F.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heel, A.J.; De Jong, A.; Song, C.; Viel, J.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Ferrario, C.; Viappiani, A.; Mancabelli, L.; Mangifesta, M.; Taminiau, B.; Delcenserie, V.; et al. Investigation of the evolutionary development of the genus Bifidobacterium by comparative genomics. Appl. Environ. Microbiol. 2014, 80, 6383–6394. [Google Scholar] [CrossRef] [Green Version]
- Ruas-Madiedo, P.; Moreno, J.A.; Salazar, N.; Delgado, S.; Mayo, B.; Margolles, A.; Reyes-Gavilán, C.G.D.L. Screening of exopolysaccharide-producing lactobacillus and Bifidobacterium strains isolated from the human intestinal microbiota. Appl. Environ. Microbiol. 2007, 73, 4385–4388. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Cantabrana, C.; Sánchez, B.; Milani, C.; Ventura, M.; Margolles, A.; Ruas-Madiedo, P. Genomic overview and biological functions of exopolysaccharide biosynthesis in Bifidobacterium spp. Appl. Environ. Microbiol. 2014, 80, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmann, F.; Kosma, P.; O’Callaghan, A.; Leahy, S.; Bottacini, F.; Molloy, E.; Plattner, S.; Schiavi, E.; Gleinser, M.; Groeger, D.; et al. Genome analysis and characterisation of the exopolysaccharide produced by Bifidobacterium longum subsp. longum 35624™. PLoS ONE 2016, 11, e0162983. [Google Scholar] [CrossRef] [Green Version]
- Figueras, M.J.; Beaz-Hidalgo, R.; Hossain, M.J.; Liles, M.R. Taxonomic affiliation of new genomes should be verified using average nucleotide identity and multilocus phylogenetic analysis. Genome Announc. 2014, 2, e00927-14. [Google Scholar] [CrossRef] [Green Version]
- LoCascio, R.G.; Desai, P.; Sela, D.A.; Weimer, B.; Mills, D.A. Broad conservation of milk utilization genes in Bifidobacterium longum subsp. infantis as revealed by comparative genomic hybridization. Appl. Environ. Microbiol. 2010, 76, 7373–7381. [Google Scholar] [CrossRef] [Green Version]
- Odamaki, T.; Bottacini, F.; Kato, K.; Mitsuyama, E.; Yoshida, K.; Horigome, A.; Xiao, J.-Z.; Van Sinderen, D. Genomic diversity and distribution of Bifidobacterium longum subsp. longum across the human lifespan. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of actinobacteria: Tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef] [Green Version]
- Locascio, R.G.; Niñonuevo, M.R.; Kronewitter, S.R.; Freeman, S.L.; German, J.B.; Lebrilla, C.B.; Mills, D.A. A versatile and scalable strategy for glycoprofiling Bifidobacterial consumption of human milk oligosaccharides. Microb. Biotechnol. 2008, 2, 333–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asakuma, S.; Hatakeyama, E.; Urashima, T.; Yoshida, E.; Katayama, T.; Yamamoto, K.; Kumagai, H.; Ashida, H.; Hirose, J.; Kitaoka, M. Physiology of consumption of human milk oligosaccharides by infant gut-associated Bifidobacteria. J. Biol. Chem. 2011, 286, 34583–34592. [Google Scholar] [CrossRef] [Green Version]
- Garrido, D.; Ruiz-Moyano, S.; Lemay, D.; Sela, D.A.; German, J.B.; Mills, D.A. Comparative transcriptomics reveals key differences in the response to milk oligosaccharides of infant gut-associated Bifidobacteria. Sci. Rep. 2015, 5, 13517. [Google Scholar] [CrossRef] [PubMed]
- Garrido, D.; Ruiz-Moyano, S.; Kirmiz, N.; Davis, J.C.; Totten, S.M.; Lemay, D.; Ugalde, J.A.; German, J.B.; Lebrilla, C.B.; Mills, D.A. A novel gene cluster allows preferential utilization of fucosylated milk oligosaccharides in Bifidobacterium longum subsp. longum SC596. Sci. Rep. 2016, 6, 35045. [Google Scholar] [CrossRef] [Green Version]
- Bunesova, V.; Lacroix, C.; Schwab, C. Fucosyllactose and L-fucose utilization of infant Bifidobacterium longum and Bifidobacterium kashiwanohense. BMC Microbiol. 2016, 16, 248. [Google Scholar] [CrossRef] [Green Version]
- Lawley, B.; Centanni, M.; Watanabe, J.; Sims, I.; Carnachan, S.; Broadbent, R.; Lee, P.S.; Wong, K.H.; Tannock, G.W. Tuf gene sequence variation in Bifidobacterium longum subsp. infantis detected in the fecal microbiota of Chinese infants. Appl. Environ. Microbiol. 2018, 84, e00336-18. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, E.; Sakurama, H.; Kiyohara, M.; Nakajima, M.; Kitaoka, M.; Ashida, H.; Hirose, J.; Katayama, T.; Yamamoto, K.; Kumagai, H. Bifidobacterium longum subsp. infantis uses two different β-galactosidases for selectively degrading type-1 and type-2 human milk oligosaccharides. Glycobiology 2012, 22, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crociani, F.; Alessandrini, A.; Mucci, M.M.; Biavati, B. Degradation of complex carbohydrates by Bifidobacterium spp. Int. J. Food Microbiol. 1994, 24, 199–210. [Google Scholar] [CrossRef]
- Sela, D.A.; Mills, D.A. Nursing our microbiota: Molecular linkages between Bifidobacteria and milk oligosaccharides. Trends Microbiol. 2010, 18, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujawska, M.; La Rosa, S.L.; Roger, L.C.; Pope, P.B.; Hoyles, L.; McCartney, A.L.; Hall, L.J. Succession of Bifidobacterium longum strains in response to a changing early life nutritional environment reveals dietary substrate adaptations. iScience 2020, 23, 101368. [Google Scholar] [CrossRef]
- Schiavi, E.; Gleinser, M.; Molloy, E.; Groeger, D.; Frei, R.; Ferstl, R.; Rodriguez-Perez, N.; Ziegler, M.; Grant, R.; Moriarty, T.F.; et al. The surface-associated exopolysaccharide of Bifidobacterium longum 35624 plays an essential role in dampening host proinflammatory responses and repressing local TH17 responses. Appl. Environ. Microbiol. 2016, 82, 7185–7196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Yang, B.; Zhao, J.; Zhao, J.; Stanton, C.; Ross, R.; Zhang, H.; Chen, W. A ropy exopolysaccharide producing strain Bifidobacterium longum subsp. longum YS108R alleviates DSS-induced colitis by maintenance of the mucosal barrier and gut microbiota modulation. Food Funct. 2019, 10, 1595–1608. [Google Scholar] [CrossRef]
- Yan, S.; Zhao, G.; Liu, X.; Zhao, J.; Zhang, H.; Chen, W. Production of exopolysaccharide by Bifidobacterium longum isolated from elderly and infant feces and analysis of priming glycosyltransferase genes. RSC Adv. 2017, 7, 31736–31744. [Google Scholar] [CrossRef] [Green Version]
- Cotter, P.D.; Ross, R.; Hill, C. Bacteriocins—A viable alternative to antibiotics? Nat. Rev. Genet. 2013, 11, 95–105. [Google Scholar] [CrossRef]
- Lee, J.-H.; Li, X.; O’Sullivan, D.J. Transcription analysis of a lantibiotic gene cluster from Bifidobacterium longum DJO10A. Appl. Environ. Microbiol. 2011, 77, 5879–5887. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.C.; Balciunas, E.M.; Converti, A.; Cotter, P.D.; Oliveira, R.P.D.S. Bacteriocin production by Bifidobacterium spp. A review. Biotechnol. Adv. 2013, 31, 482–488. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genome Size (Mbp) | GC (%) | CDS Number | Origin | Accession Number |
|---|---|---|---|---|---|
| 2 | 2.62 | 59.40 | 2075 | Human Faeces | SAMEA5770183 |
| 4 | 2.57 | 59.60 | 2053 | Human Faeces | SAMEA5770185 |
| 6 | 2.83 | 59.90 | 2405 | Human Faeces | SAMEA5770187 |
| 105-A | 2.29 | 60.10 | 1770 | Human Faeces | SAMD00019943 |
| 157F | 2.41 | 60.11 | 1929 | Infant Faeces | SAMD00060953 |
| 17-1B | 2.47 | 60.20 | 1959 | Human Faeces | SAMN02862993 |
| 1888B | 2.58 | 59.40 | 2039 | Infant Faeces | SAMN06621708 |
| 35624 | 2.26 | 60.00 | 1758 | Human Faeces | SAMN04254466 |
| 35B | 2.51 | 60.10 | 1967 | Human Faeces | SAMN00829158 |
| 379 | 2.39 | 60.20 | 1902 | Human Faeces | SAMN04155602 |
| AF05-2 | 2.43 | 60.40 | 1978 | Human Faeces | SAMN09734186 |
| AH1206 | 2.42 | 60.20 | 1955 | Infant Faeces | SAMN04576213 |
| APC1480 | 2.48 | 59.90 | 2017 | Human Faeces | SAMN07958358 |
| ATCC15697 | 2.83 | 59.90 | 2411 | Infant Faeces | SAMN02598380 |
| ATCC55813 | 2.40 | 60.10 | 1901 | Infant Faeces | SAMN00001475 |
| BAMA-B05 | 2.27 | 59.90 | 1800 | Human Faeces | SAMN12569298 |
| BBMN68 | 2.27 | 59.90 | 1747 | Human Faeces | SAMN02603469 |
| BG7 | 2.46 | 60.01 | 1926 | Infant Faeces | SAMN03271682 |
| Bi-26 | 2.61 | 59.30 | 2078 | Infant Faeces | SAMN10380491 |
| Bifido_04 | 2.58 | 59.70 | 2110 | Human Blood | SAMEA51817918 |
| BI-G201 | 2.57 | 59.30 | 2028 | Human Faeces | SAMN14908987 |
| BIO5478 | 2.57 | 59.40 | 2093 | Infant Faeces | SAMN12856543 |
| BT1 | 2.58 | 59.40 | 2030 | Infant Faeces | SAMN03271683 |
| C1A13 | 2.31 | 59.86 | 2000 | Human Faeces | SAMN19128425 |
| C6A3 | 2.40 | 60.03 | 2037 | Human Faeces | SAMN16976870 |
| CCFM685 | 2.35 | 59.69 | 2025 | Infant Faeces | SAMN16976872 |
| CCFM686 | 2.50 | 59.76 | 2125 | Infant Faeces | SAMN19128426 |
| CCFM752 | 2.28 | 59.70 | 1939 | Infant Faeces | SAMN17575072 |
| CCFM756 | 2.34 | 60.00 | 2075 | Infant Faeces | SAMN16976873 |
| CCFM762 | 2.40 | 59.89 | 2037 | Infant Faeces | SAMN19128427 |
| CECT7347 | 2.33 | 60.00 | 1869 | Unknown | SAMEA3146249 |
| DJO10A | 2.39 | 60.12 | 1874 | Unknown | SAMN02603512 |
| EK3 | 2.56 | 59.40 | 2061 | Human Faeces | SAMN02862995 |
| FGXBM14M6 | 2.52 | 60.15 | 2262 | Human Faeces | SAMN16976885 |
| FGZ17I1M1 | 2.72 | 59.60 | 2583 | Infant Faeces | SAMN16976996 |
| FGZ19I1M3 | 2.66 | 59.66 | 2506 | Infant Faeces | SAMN16976991 |
| FGZ19I2M3 | 2.72 | 59.60 | 2574 | Infant Faeces | SAMN16976986 |
| FGZ23I1M2 | 2.72 | 59.60 | 2597 | Infant Faeces | SAMN16976987 |
| FHeJZ25M6 | 2.50 | 59.96 | 2150 | Human Faeces | SAMN19128429 |
| FHeJZ28M10 | 2.39 | 59.99 | 2033 | Human Faeces | SAMN19128430 |
| FHeJZ44M8 | 2.72 | 59.22 | 2551 | Infant Faeces | SAMN16976998 |
| FHeNJZ3M1 | 2.71 | 59.70 | 2599 | Infant Faeces | SAMN16976982 |
| FHNFQ45M2 | 2.84 | 59.57 | 2781 | Infant Faeces | SAMN16976993 |
| FHuBZX17M2 | 2.43 | 60.14 | 2122 | Human Faeces | SAMN19128431 |
| FHuNCS6M8 | 2.55 | 59.40 | 2332 | Infant Faeces | SAMN16976988 |
| FJND16M4 | 2.56 | 59.39 | 2390 | Infant Faeces | SAMN16976985 |
| FJND2M2 | 2.54 | 59.51 | 2296 | Infant Faeces | SAMN16976992 |
| FJSYZ1M3 | 2.63 | 59.70 | 2441 | Infant Faeces | SAMN16976990 |
| FSCREG2M33 | 2.36 | 60.16 | 2030 | Human Faeces | SAMN16976909 |
| FZJJH13M4 | 2.57 | 59.62 | 2336 | Infant Faeces | SAMN19128438 |
| FZXDX6M32 | 2.31 | 59.77 | 1961 | Human Faeces | SAMN16976921 |
| GT15 | 2.34 | 60.00 | 1815 | Human Faeces | SAMN03093230 |
| GX17A9 | 2.49 | 59.88 | 2072 | Human Faeces | SAMN16976922 |
| HeNJZ8M1 | 2.67 | 59.55 | 2447 | Infant Faeces | SAMN16976984 |
| HUB3617 | 2.46 | 59.94 | 2175 | Human Faeces | SAMN19128434 |
| IN-07 | 2.75 | 60.00 | 2189 | Human Faeces | SAMD00047616 |
| IN-F29 | 2.64 | 59.90 | 2109 | Human Faeces | SAMD00047617 |
| JSSZ7M7 | 2.62 | 59.35 | 2330 | Infant Faeces | SAMN16976979 |
| JSWX23M3 | 2.56 | 59.38 | 2311 | Infant Faeces | SAMN19128437 |
| JSWX25M6 | 2.62 | 59.55 | 2372 | Infant Faeces | SAMN16976983 |
| JSWX3M1 | 2.63 | 58.89 | 2306 | Infant Faeces | SAMN16976989 |
| JSWX6M2 | 2.72 | 59.92 | 2569 | Infant Faeces | SAMN16976980 |
| JSWX9M5 | 2.54 | 60.04 | 2276 | Infant Faeces | SAMN19128439 |
| LH23 | 2.76 | 59.60 | 2293 | Infant Faeces | SAMEA4838174 |
| LH665 | 2.59 | 59.40 | 2087 | Infant Faeces | SAMEA4838176 |
| M120R013 | 2.20 | 60.02 | 1897 | Human Faeces | SAMN16976924 |
| M203F0227 | 2.77 | 59.61 | 2639 | Infant Faeces | SAMN16976997 |
| M2CF0114 | 2.37 | 59.77 | 2085 | Human Faeces | SAMN19128435 |
| MC2 | 2.56 | 59.60 | 2011 | Human Faeces | SAMEA5574696 |
| MC-42 | 2.29 | 59.80 | 1775 | Infant Faeces | SAMN04263942 |
| NCC2705 | 2.26 | 60.11 | 1769 | Infant Faeces | SAMN02603675 |
| NCTC13219 | 2.60 | 60.00 | 2235 | Unknown | SAMEA104318167 |
| PC1 | 2.79 | 59.80 | 2387 | Human Faeces | SAMEA51825418 |
| RG23 | 2.26 | 60.02 | 1911 | Human Faeces | SAMN16976925 |
| RG41 | 2.49 | 60.25 | 2147 | Human Faeces | SAMN19128436 |
| SDZC2M4 | 2.73 | 59.64 | 2586 | Infant Faeces | SAMN16976981 |
| TPY12-1 | 2.64 | 59.70 | 2128 | Infant Faeces | SAMN05578879 |
| UBBI-01 | 2.73 | 59.40 | 2234 | Fermented Food | SAMN11370925 |
| UMB0788 | 2.45 | 60.20 | 2012 | Female Urinary Tract | SAMN08193649 |
| YS108R | 2.52 | 60.10 | 2021 | Human Faeces | SAMN09355369 |
| Enzymes/Carbohydrates | Gene (Cluster) | B. longum subsp. longum | B. longum subsp. infantis |
|---|---|---|---|
| Fucosidase | BLON_RS01290- BLON_RS01310 | Absent | Present (except FHeNJZ3M1, SDZC2M4) |
| Fucosidase | BLON_RS02195-BLON_RS02205 | Absent | Present (except FHeNJZ3M1, SDZC2M4) |
| Sialidases | BLON_RS03255-BLON_RS03305 | Absent | Present |
| beta-galactosidases | BLON_RS10470 | Present | Present |
| N-acetyl-β-D-hexosaminidases | BLON_RS02365 | Absent (except JSWX9M5) | Present (except LH665) |
| N-acetyl-β-D-hexosaminidases | BLON_RS03705 | Present | Present |
| LNB | BLON_RS11215- BLON_RS11245 | Present (except M2CF0114) | Present |
| Gene ID | Gene Name | Protein ID | Annotation |
|---|---|---|---|
| BL_RS05080 | araA | WP_007051388.1 | L-arabinose isomerase |
| BL_RS05085 | araB | WP_007051389.1 | L-ribulose-5-phosphate 4-epimerase |
| BL_RS05090 | araD | WP_007051390.1 | FGGY-family carbohydrate kinase |
| BL_RS05095 | - | WP_008782880.1 | LacI family DNA-binding transcriptional regulator |
| BL_RS05100 | - | WP_011068401.1 | ribonuclease HII |
| BL_RS05105 | lepB | WP_010081174.1 | signal peptidase I |
| Bacteriocin | B. longum subsp. longum | B. longum subsp. infantis | Class | Sources |
|---|---|---|---|---|
| Propionicin_SM1 | 1 | 26 | II | Propionibacterium jensenii |
| Michiganin-A | - | 18 | Ia, Lanthipeptide B | Clavibacter michiganensis subsp. michiganensis |
| Geobacillin_I_like | - | 12 | Ia, Lanthipeptide A | Geobacillus kaustophilus HTA426 |
| BLD_1648 | 3 | 19 | Ia, Lanthipeptide B | B. longum subsp. longum DJO10A |
| Propeptin_2 | - | 4 | If, Lasso_peptide | Microbispora sp. SNA-115 |
| Nai_112 | - | 2 | Ia, Lanthipeptide C | Actinoplanes sp. NAI112 |
| Variacin | - | 4 | Ia, Lanthipeptide B | kocuria varians |
| Planosporicin | - | 4 | Ia, Lanthipeptide A | Microbispora sp. 107891 |
| Flavucin | - | 3 | Ia, Lanthipeptide A | Corynebacterium lipophiloflavum |
| Thiopeptide | - | 4 | Thiopeptide | - |
| LAPs | - | 5 | Id, LAPs | - |
| Sactipeptides | - | 3 | Ic, Sactipeptides | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Zhou, X.; Stanton, C.; Ross, R.P.; Zhao, J.; Zhang, H.; Yang, B.; Chen, W. Comparative Genomics Analyses Reveal the Differences between B. longum subsp. infantis and B. longum subsp. longum in Carbohydrate Utilisation, CRISPR-Cas Systems and Bacteriocin Operons. Microorganisms 2021, 9, 1713. https://doi.org/10.3390/microorganisms9081713
Li M, Zhou X, Stanton C, Ross RP, Zhao J, Zhang H, Yang B, Chen W. Comparative Genomics Analyses Reveal the Differences between B. longum subsp. infantis and B. longum subsp. longum in Carbohydrate Utilisation, CRISPR-Cas Systems and Bacteriocin Operons. Microorganisms. 2021; 9(8):1713. https://doi.org/10.3390/microorganisms9081713
Chicago/Turabian StyleLi, Mingjie, Xingya Zhou, Catherine Stanton, R. Paul Ross, Jianxin Zhao, Hao Zhang, Bo Yang, and Wei Chen. 2021. "Comparative Genomics Analyses Reveal the Differences between B. longum subsp. infantis and B. longum subsp. longum in Carbohydrate Utilisation, CRISPR-Cas Systems and Bacteriocin Operons" Microorganisms 9, no. 8: 1713. https://doi.org/10.3390/microorganisms9081713
APA StyleLi, M., Zhou, X., Stanton, C., Ross, R. P., Zhao, J., Zhang, H., Yang, B., & Chen, W. (2021). Comparative Genomics Analyses Reveal the Differences between B. longum subsp. infantis and B. longum subsp. longum in Carbohydrate Utilisation, CRISPR-Cas Systems and Bacteriocin Operons. Microorganisms, 9(8), 1713. https://doi.org/10.3390/microorganisms9081713