
Differences in Gut Virome Related to Barrett Esophagus and Esophageal Adenocarcinoma

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Virome DNA Extraction
2.3. Bioinformatic Analysis
2.4. Statistics Analysis
3. Results
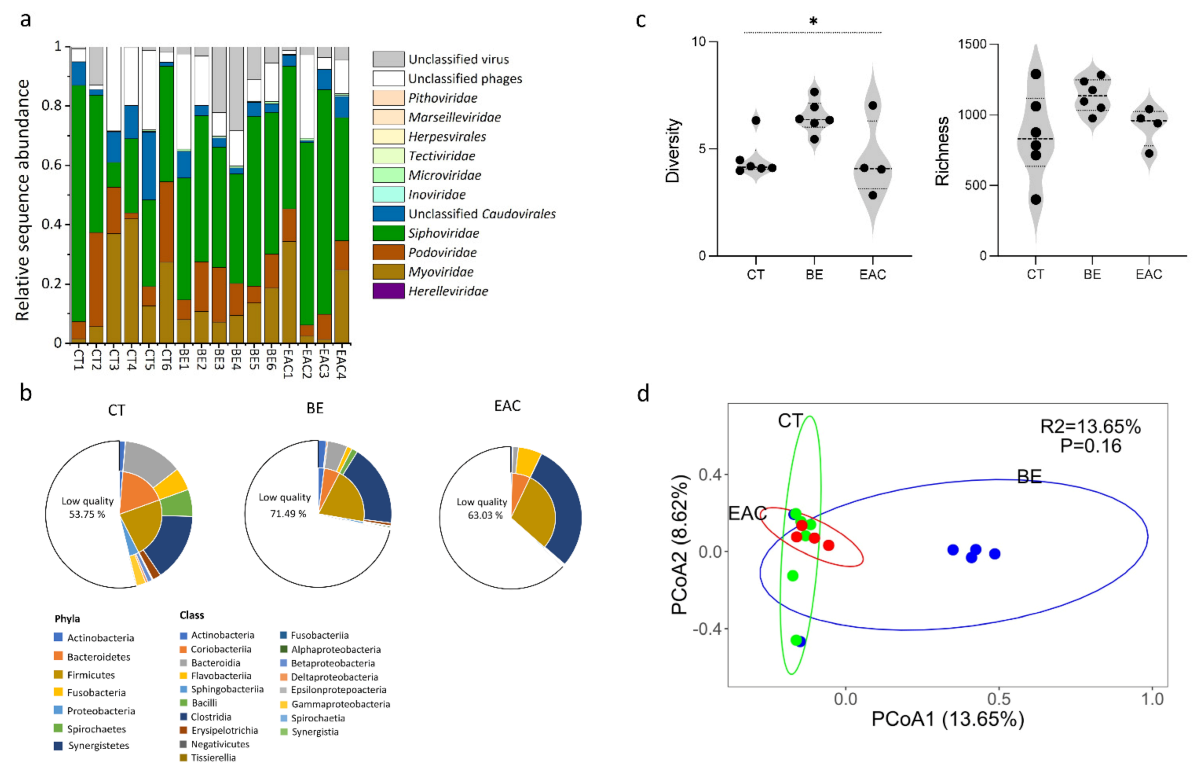
3.1. Gut Bacteriophage Community Structure Differed for BE and EAC Compared to Their Healthy Counterparts
3.2. Abundant and Rare Phage Communities in the Gut May Contribute to the Progress of Esophageal Carcinogesis
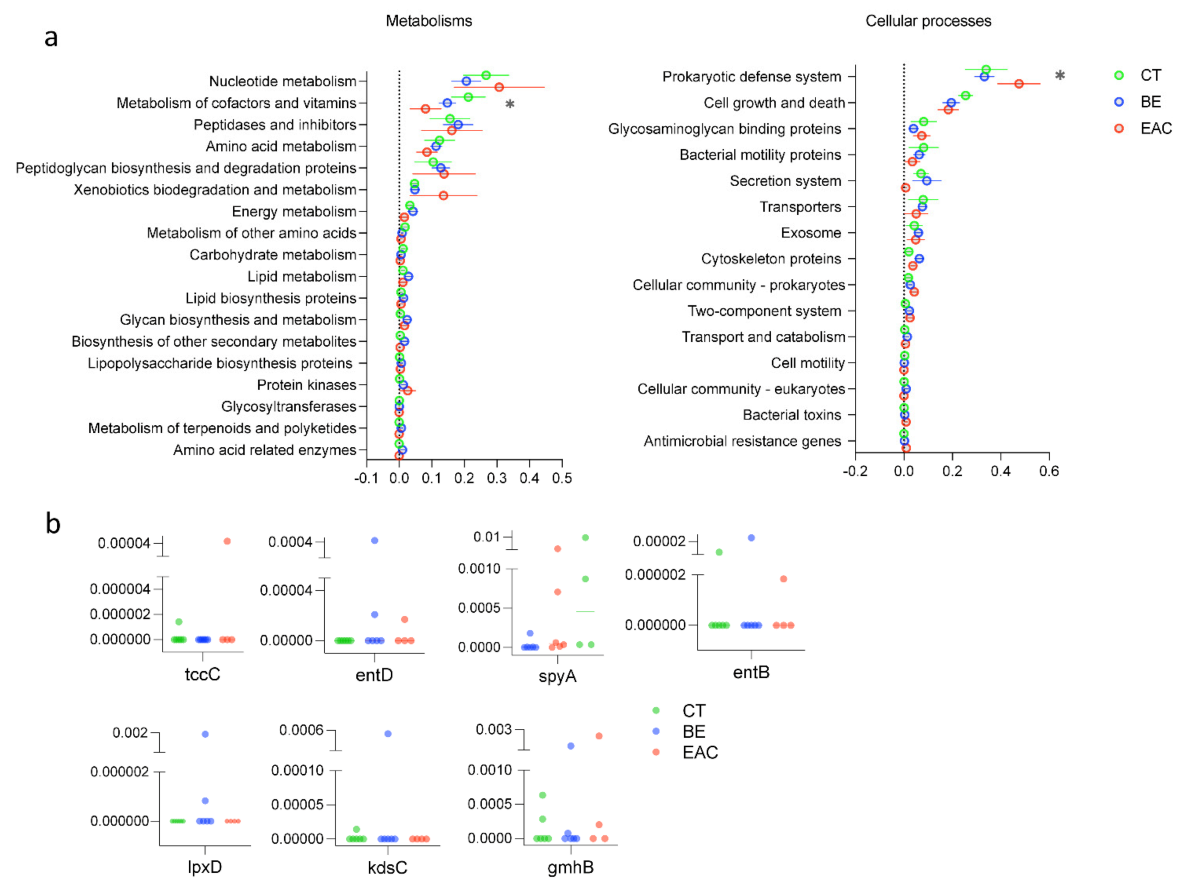
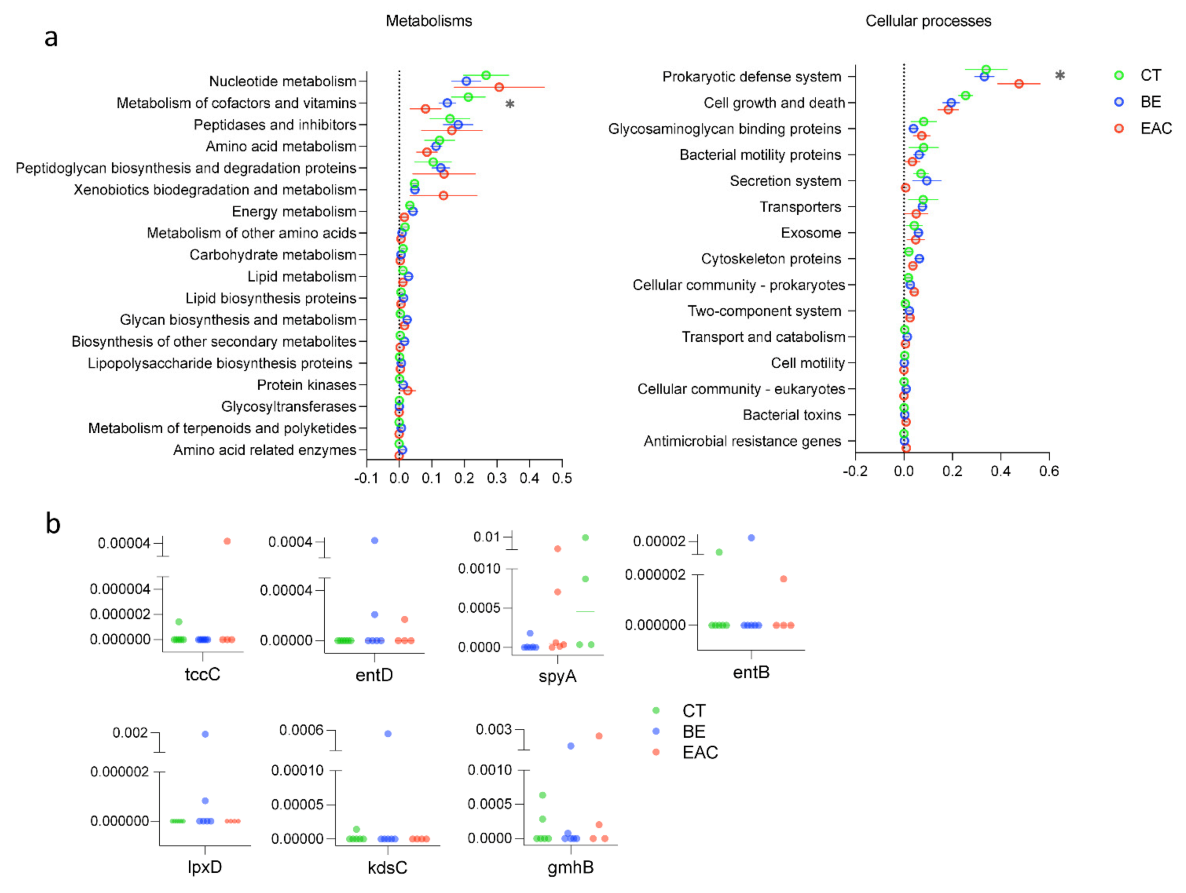
3.3. AMGs Found in Rare Bacteriophages Showed Increment in Esophageal Diseases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, Y.; Li, D.; Shan, B.; Liang, D.; Shi, J.; Chen, W.; He, J. Incidence and mortality of esophagus cancer in China, 2008–2012. Chin. J. Cancer Res. 2019, 31, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Pennathur, A.; Gibson, M.K.; Jobe, B.A.; Luketich, J.D. Oesophageal carcinoma. Lancet 2013, 381, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Martinucci, I.; De Bortoli, N.; Russo, S.; Bertani, L.; Furnari, M.; Mokrowiecka, A.; Malecka-Panas, E.; Savarino, V.; Savarino, E.; Marchi, S. Barrett’s esophagus in 2016: From pathophysiology to treatment. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 190–206. [Google Scholar] [CrossRef]
- Pohl, H.; Koch, M.; Khalifa, A.; Papanikolaou, I.; Scheiner, K.; Wiedenmann, B.; Rösch, T. Evaluation of endocytoscopy in the surveillance of patients with Barrett’s esophagus. Endoscopy 2007, 39, 492–496. [Google Scholar] [CrossRef]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef]
- Münch, N.S.; Fang, H.-Y.; Ingermann, J.; Maurer, H.C.; Anand, A.; Kellner, V.; Sahm, V.; Wiethaler, M.; Baumeister, T.; Wein, F.; et al. High-fat diet accelerates carcinogenesis in a mouse model of barrett’s esophagus via interleukin 8 and alterations to the gut microbiome. Gastroenterology 2019, 157, 492–506. [Google Scholar] [CrossRef] [Green Version]
- Snider, E.J.; Compres, G.; Freedberg, D.E.; Khiabanian, H.; Nobel, Y.R.; Stump, S.; Uhlemann, A.-C.; Lightdale, C.J.; Abrams, J.A. Alterations to the esophageal microbiome associated with progression from Barrett’s esophagus to esophageal adenocarcinoma. Cancer Epidemiol. Prev. Biomark. 2019, 28, 1687–1693. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef]
- Manor, O.; Dai, C.L.; Kornilov, S.A.; Smith, B.; Price, N.D.; Lovejoy, J.C.; Gibbons, S.M.; Magis, A.T. Health and disease markers correlate with gut microbiome composition across thousands of people. Nat. Commun. 2020, 11, 5206. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Francois, F.; Pei, Z. Molecular pathways: Pathogenesis and clinical implications of microbiome alteration in esophagitis and Barrett esophagus. Clin. Cancer Res. 2012, 18, 2138–2144. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, A.H.; Kelly, L.A.; Kreft, R.E.; Barlek, M.; Omstead, A.N.; Matsui, D.; Boyd, N.H.; Gazarik, K.E.; Heit, M.I.; Nistico, L.; et al. Associations of microbiota and toll-like receptor signaling pathway in esophageal adenocarcinoma. BMC Cancer 2016, 16, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okereke, I.; Hamilton, C.; Wenholz, A.; Jala, V.; Giang, T.; Reynolds, S.; Miller, A.; Pyles, R. Associations of the microbiome and esophageal disease. J. Thorac. Dis. 2019, 11, S1588–S1593. [Google Scholar] [CrossRef]
- Wylie, K.M.; Weinstock, G.M.; Storch, G.A. Emerging view of the human virome. Transl. Res. 2012, 160, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carding, S.R.; Davis, N.; Hoyles, L. The human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K. The virome in host health and disease. Immunity 2015, 42, 805–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepage, P.; Leclerc, M.C.; Joossens, M.; Mondot, S.; Blottière, H.M.; Raes, J.; Ehrlich, D.; Doré, J. A metagenomic insight into our gut’s microbiome. Gut 2013, 62, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.; Shanahan, F.; Stanton, C.; Hill, C.; Coffey, A.; Ross, R.P. Movers and shakers: Influence of bacteriophages in shaping the mammalian gut microbiota. Gut Microbes 2013, 4, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, M.; Hill, C.; Ross, R.P. Exploiting gut bacteriophages for human health. Trends Microbiol. 2014, 22, 399–405. [Google Scholar] [CrossRef]
- Ungaro, F.; Massimino, L.; Furfaro, F.; Rimoldi, V.; Peyrin-Biroulet, L.; D’alessio, S.; Danese, S. Metagenomic analysis of intestinal mucosa revealed a specific eukaryotic gut virome signature in early-diagnosed inflammatory bowel disease. Gut Microbes 2019, 10, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Tetz, G.; Tetz, V. Prion-like domains in eukaryotic viruses. Sci. Rep. 2018, 8, 8931. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Sabino, J.; Hirten, R.P.; Colombel, J.F. bacteriophages in gastroenterology—From biology to clinical applications. Aliment. Pharmacol. Ther. 2020, 51, 53–63. [Google Scholar] [CrossRef]
- De Sordi, L.; Lourenço, M.; Debarbieux, L. The battle within: Interactions of bacteriophages and bacteria in the gastrointestinal tract. Cell Host Microbe 2019, 25, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Jończyk-Matysiak, E.; Wojciechowska, R.; Górski, A. Bacteriophages in the gastrointestinal tract and their implications. Gut Pathog. 2017, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaei, M.K.; Xue, J.; Costa, R.; Ru, J.; Schulz, S.; Taranu, Z.E.; Deng, L. Challenges of studying the human virome–relevant emerging technologies. Trends Microbiol. 2021, 29, 171–181. [Google Scholar] [CrossRef]
- Lin, E.C.; Lynch, A.S. Regulation of Gene Expression in Escherichia coli; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Feiner, R.; Argov, T.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. A new perspective on lysogeny: Prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 2015, 13, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, J.R.; Allison, H.; James, C.E.; McCarthy, A.J.; Sharp, R. Phage-mediated transfer of virulence genes. J. Chem. Technol. Biotechnol. 2001, 76, 662–666. [Google Scholar] [CrossRef]
- Coleman, D.C.; Sullivan, D.J.; Russel, R.J.; Arbuthnott, J.P.; Carey, B.F.; Pomeroy, H.M. Staphylococcus aureus bacteriophages mediating the simultaneous lysogenic conversion of β-lysin, staphylokinase and enterotoxin A: Molecular mechanism of triple conversion. Microbiology 1989, 135, 1679–1697. [Google Scholar] [CrossRef] [Green Version]
- Crummett, L.T.; Puxty, R.J.; Weihe, C.; Marston, M.F.; Martiny, J.B. The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology 2016, 499, 219–229. [Google Scholar] [CrossRef]
- Tetz, G.; Brown, S.M.; Hao, Y.; Tetz, V. Parkinson’s disease and bacteriophages as its overlooked contributors. Sci. Rep. 2018, 8, 10812. [Google Scholar] [CrossRef] [Green Version]
- Tetz, G.; Brown, S.M.; Hao, Y.; Tetz, V. Type 1 diabetes: An association between autoimmunity, the dynamics of gut amyloid-producing E. coli and their phages. Sci. Rep. 2019, 9, 9685. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.-H.; Sugimura, N.; Lam, T.Y.-T.; et al. Alterations in enteric virome are associated with colorectal cancer and survival outcomes. Gastroenterology 2018, 155, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerkop, B.A.; Kleiner, M.; Paez-Espino, D.; Zhu, W.; Bushnell, B.; Hassell, B.; Winter, S.E.; Kyrpides, N.C.; Hooper, L.V. Murine colitis reveals a disease-associated bacteriophage community. Nat. Microbiol. 2018, 3, 1023–1031. [Google Scholar] [CrossRef]
- Ly, M.; Abeles, S.R.; Boehm, T.K.; Robles-Sikisaka, R.; Naidu, M.; Santiago-Rodriguez, T.; Pride, D.T. Altered oral viral ecology in association with periodontal disease. MBio 2014, 5, e01133-14. [Google Scholar] [CrossRef] [Green Version]
- Abeles, S.R.; Robles-Sikisaka, R.; Ly, M.; Lum, A.G.; Salzman, J.; Boehm, T.K.; Pride, D.T. Human oral viruses are personal, persistent and gender-consistent. ISME J. 2014, 8, 1753–1767. [Google Scholar] [CrossRef]
- Deshpande, N.P.; Riordan, S.M.; Castaño-Rodríguez, N.; Wilkins, M.R.; Kaakoush, N.O. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome 2018, 6, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiethaler, M.; Slotta-Huspenina, J.; Brandtner, A.; Horstmann, J.; Wein, F.; Baumeister, T.; Radani, N.; Gerland, S.; Anand, A.; Lange, S.; et al. BarrettNET—A prospective registry for risk estimation of patients with Barrett’s esophagus to progress to adenocarcinoma. Dis. Esophagus 2019, 32, doz024. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- von Meijenfeldt, F.B.; Arkhipova, K.; Cambuy, D.D.; Coutinho, F.H.; Dutilh, B.E. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 2019, 20, 217. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying viruses from metagenomic data using deep learning. Quant. Biol. 2020, 8, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.B.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ren, J.; Tang, K.; Dart, E.; Ignacio-Espinoza, J.C.; Fuhrman, J.A.; Braun, J.; Sun, F.; Ahlgren, N.A. A network-based integrated framework for predicting virus–prokaryote interactions. NAR Genom. Bioinform. 2020, 2, lqaa044. [Google Scholar] [CrossRef]
- Ji, M.; Kong, W.; Stegen, J.; Yue, L.; Wang, F.; Dong, X.; Cowan, D.A.; Ferrari, B.C. Distinct assembly mechanisms underlie similar biogeographical patterns of rare and abundant bacteria in Tibetan Plateau grassland soils. Environ. Microbiol. 2020, 22, 2261–2272. [Google Scholar] [CrossRef]
- Sjöstedt, J.; Koch-Schmidt, P.; Pontarp, M.; Canbäck, B.; Tunlid, A.; Lundberg, P.; Hagström, Å.; Riemann, L. Recruitment of members from the rare biosphere of marine bacterioplankton communities after an environmental disturbance. Appl. Environ. Microbiol. 2012, 78, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, A.; Cruaud, P.; Alsop, E.; de Rezende, J.R.; Head, I.M.; Tsesmetzis, N. Beyond the tip of the iceberg; a new view of the diversity of sulfite-and sulfate-reducing microorganisms. ISME J. 2018, 12, 2096–2099. [Google Scholar] [CrossRef]
- Coye, L.H.; Collins, C.M. Identification of SpyA, a novel ADP-ribosyltransferase of Streptococcus pyogenes. Mol. Microbiol. 2004, 54, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Leidreiter, F.; Roderer, D.; Meusch, D.; Gatsogiannis, C.; Benz, R.; Raunser, S. Common architecture of Tc toxins from human and insect pathogenic bacteria. Sci. Adv. 2019, 5, eaax6497. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Tang, D.; Hou, P.; Shen, W.; Li, H.; Wang, T.; Liu, R. Dysbiosis of gut microbiota in patients with esophageal cancer. Microb. Pathog. 2021, 150, 104709. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Wang, X.; Gai, Z.; Song, X.; Jia, X.; Tian, H. Bile acids but not acidic acids induce Barrett’s esophagus. Int. J. Clin. Exp. Pathol. 2015, 8, 1384. [Google Scholar] [PubMed]
- Schmidt, M.; Ankerst, D.P.; Chen, Y.; Wiethaler, M.; Slotta-Huspenina, J.; Becker, K.-F.; Horstmann, J.; Kohlmayer, F.; Lehmann, A.; Linkohr, B.; et al. Epidemiologic Risk Factors in a Comparison of a Barrett Esophagus Registry (BarrettNET) and a Case–Control Population in Germany. Cancer Prev. Res. 2020, 13, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Elliott, D.R.F.; Perner, J.; Li, X.; Symmons, M.F.; Verstak, B.; Eldridge, M.; Bower, L.; O’Donovan, M.; Gay, N.J.; Fitzgerald, R.C. Impact of mutations in Toll-like receptor pathway genes on esophageal carcinogenesis. PLoS Genet. 2017, 13, e1006808. [Google Scholar]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T.; Koumpouras, C.C.; Schloss, P.D. Diagnostic potential and interactive dynamics of the colorectal cancer virome. MBio 2018, 9, e02248-18. [Google Scholar] [CrossRef] [Green Version]
- Sinha, A.; Maurice, C.F. Bacteriophages: Uncharacterized and dynamic regulators of the immune system. Mediat. Inflamm. 2019, 2019, 3730519. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Baker, K.; Padman, B.S.; Patwa, R.; Dunstan, R.A.; Weston, T.A.; Schlosser, K.; Bailey, B.; Lithgow, T.; Lazarou, M.; et al. Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. MBio 2017, 8, e01874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bichet, M.C.; Chin, W.H.; Richards, W.; Lin, Y.-W.; Avellaneda-Franco, L.; Hernandez, C.A.; Oddo, A.; Chernyavskiy, O.; Hilsenstein, V.; Neild, A. Bacteriophage uptake by mammalian cell layers represents a potential sink that may impact phage therapy. Iscience 2021, 24, 102287. [Google Scholar] [CrossRef]
- Wahida, A.; Tang, F.; Barr, J.J. Rethinking phage-bacteria-eukaryotic relationships and their influence on human health. Cell Host Microbe 2021, 29, 681–688. [Google Scholar] [CrossRef]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of bacteriophages is linked to aggravated intestinal inflammation and colitis. Cell Host Microbe 2019, 25, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Moonen, A.; Annese, V.; Belmans, A.; Bredenoord, A.J.; Des Varannes, S.B.; Costantini, M.; Dousset, B.; Elizalde, J.I.; Fumagalli, U.; Gaudric, M.; et al. Long-term results of the European achalasia trial: A multicentre randomised controlled trial comparing pneumatic dilation versus laparoscopic Heller myotomy. Gut 2016, 65, 732–739. [Google Scholar] [CrossRef]
- Pei, Z.; Bini, E.J.; Yang, L.; Zhou, M.; Francois, F.; Blaser, M.J. Bacterial biota in the human distal esophagus. Proc. Natl. Acad. Sci. USA 2004, 101, 4250–4255. [Google Scholar] [CrossRef] [Green Version]
- Pei, Z.; Yang, L. Bacterial biota in reflux esophagitis and Barrett’s esophagus. World J. Gastroenterol. 2005, 11, 7277–7283. [Google Scholar] [CrossRef] [PubMed]
- Fillon, S.A.; Harris, J.K.; Wagner, B.D.; Kelly, C.J.; Stevens, M.J.; Moore, W.; Fang, R.; Schroeder, S.; Masterson, J.C.; Robertson, C.E.; et al. Novel device to sample the esophageal microbiome—The esophageal string test. PLoS ONE 2012, 7, e42938. [Google Scholar] [CrossRef]
- Lim, E.H.; Zhang, S.-L.; Li, J.-L.; Yap, W.-S.; Howe, T.-C.; Tan, B.-P.; Lee, Y.-S.; Wong, D.; Khoo, K.-L.; Seto, K.-Y.; et al. Using whole genome amplification (WGA) of low-volume biopsies to assess the prognostic role of EGFR, KRAS, p53, and CMET mutations in advanced-stage non-small cell lung cancer (NSCLC). J. Thorac. Oncol. 2009, 4, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, M.J.; Plummer, N.T. Part 1: The human gut microbiome in health and disease. Integr. Med. A Clin. J. 2014, 13, 17. [Google Scholar]
- Clooney, A.G.; Sutton, T.D.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.J.; Draper, L.A.; Plevy, S.E.; Ross, R.P.; et al. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe 2019, 26, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Tetz, G.; Tetz, V. Bacteriophage infections of microbiota can lead to leaky gut in an experimental rodent model. Gut Pathog. 2016, 8, 33. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, T.; Ru, J.; Xue, J.; Schulz, S.; Mirzaei, M.K.; Janssen, K.-P.; Quante, M.; Deng, L. Differences in Gut Virome Related to Barrett Esophagus and Esophageal Adenocarcinoma. Microorganisms 2021, 9, 1701. https://doi.org/10.3390/microorganisms9081701
Ma T, Ru J, Xue J, Schulz S, Mirzaei MK, Janssen K-P, Quante M, Deng L. Differences in Gut Virome Related to Barrett Esophagus and Esophageal Adenocarcinoma. Microorganisms. 2021; 9(8):1701. https://doi.org/10.3390/microorganisms9081701
Chicago/Turabian StyleMa, Tianli, Jinlong Ru, Jinling Xue, Sarah Schulz, Mohammadali Khan Mirzaei, Klaus-Peter Janssen, Michael Quante, and Li Deng. 2021. "Differences in Gut Virome Related to Barrett Esophagus and Esophageal Adenocarcinoma" Microorganisms 9, no. 8: 1701. https://doi.org/10.3390/microorganisms9081701
APA StyleMa, T., Ru, J., Xue, J., Schulz, S., Mirzaei, M. K., Janssen, K.-P., Quante, M., & Deng, L. (2021). Differences in Gut Virome Related to Barrett Esophagus and Esophageal Adenocarcinoma. Microorganisms, 9(8), 1701. https://doi.org/10.3390/microorganisms9081701