
Distribution of SARS-CoV-2 Lineages in the Czech Republic, Analysis of Data from the First Year of the Pandemic

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Generation of Controls
2.3. Calculation of Ct for Samples and Controls
2.4. NGS Library Preparation
2.4.1. NGS Library Preparation—Illumina
2.4.2. NGS Library Preparation: NEBNext + Twist (NEB + TWIST) Combined Workflow
2.4.3. NGS Library Preparation-Twist Workflow
2.5. Massively Parallel Sequencing
2.6. Reference Mapping Data Analysis
2.7. De Novo Assembly Data Analysis
2.8. SARS-CoV-2 Lineage Classification
2.9. Data Availability
2.10. Ethics
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. NextStrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Forster, P.; Forster, L.; Renfrew, C.; Forster, M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 9241–9243. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.C.; Chen, C.H.; Wang, J.H.; Liao, H.C.; Yang, C.T.; Chen, C.W.; Lin, Y.C.; Kao, C.H.; Lu, M.Y.J.; Liao, J.C. Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations. Proc. Natl. Acad. Sci. USA 2020, 117, 30679–30686. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Komenda, M.; Bulhart, V.; Karolyi, M.; Jarkovský, J.; Mužík, J.; Májek, O.; Šnajdrová, L.; Růžičková, P.; Rážová, J.; Prymula, R.; et al. Complex reporting of the COVID-19 epidemic in the Czech Republic: Use of an interactive web-based app in practice. J. Med. Internet Res. 2020, 22, e19367. [Google Scholar] [CrossRef]
- Czechia: WHO Coronavirus Disease (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int (accessed on 24 July 2021).
- These European Countries Are Seeing Worse Coronavirus Spikes. Available online: https://time.com/5902172/europe-coronavirus-second-wave-belgium-czech-republic/ (accessed on 24 July 2021).
- Hasell, J.; Mathieu, E.; Beltekian, D.; Macdonald, B.; Giattino, C.; Ortiz-Ospina, E.; Roser, M.; Ritchie, H. A cross-country database of COVID-19 testing. Sci. Data 2020, 7, 345. [Google Scholar] [CrossRef] [PubMed]
- Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 24 July 2021).
- CDC. Coronavirus Disease 2019 (COVID-19). Centers for Disease Control and Prevention. 2020. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 25 July 2021).
- Li, D.; Zhang, J.; Li, J. Primer design for quantitative real-time PCR for the emerging Coronavirus SARS-CoV-2. Theranostics 2020, 10, 7150–7162. [Google Scholar] [CrossRef]
- Klempt, P.; Brož, P.; Kašný, M.; Novotný, A.; Kvapilová, K.; Kvapil, P. Performance of targeted library preparation solutions for SARS-CoV-2 whole genome analysis. Diagnostics 2020, 10, 769. [Google Scholar] [CrossRef] [PubMed]
- Kriegova, E.; Fillerova, R.; Kvapil, P. Direct-RT-qPCR Detection of SARS-CoV-2 without RNA Extraction as Part of a COVID-19 Testing Strategy: From Sample to Result in One Hour. Diagnostics 2020, 10, 605. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Popa, A.; Genger, J.-W.; Nicholson, M.D.; Penz, T.; Schmid, D.; Aberle, S.W.; Agerer, B.; Lercher, A.; Endler, L.; Colaço, H.; et al. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci. Transl. Med. 2020, 12, eabe2555. [Google Scholar] [CrossRef] [PubMed]
- Serwin, K.; Ossowski, A.; Szargut, M.; Cytacka, S.; Urbańska, A.; Majchrzak, A.; Niedźwiedź, A.; Czerska, E.; Pawińska-Matecka, A.; Gołąb, J.; et al. Molecular Evolution and Epidemiological Characteristics of SARS COV-2 in (Northwestern) Poland. Viruses 2021, 13, 1295. [Google Scholar] [CrossRef]
- World Health Organization. Clinical Management of COVID-19: Interim Guidance, 27 May 2020. Available online: https://apps.who.int/iris/handle/10665/332196 (accessed on 24 July 2021).
- Park, S.E. Epidemiology, virology, and clinical features of severe acute respiratory syndrome-coronavirus-2 (SARS-COV-2; Coronavirus Disease-19). Clin. Exp. Pediatrics 2020, 63, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef]
- du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef]
- Pedro, N.; Silva, C.N.; Magalhães, A.C.; Cavadas, B.; Rocha, A.M.; Moreira, A.C.; Gomes, M.S.; Silva, D.; Sobrinho-Simões, J.; Ramos, A.; et al. Dynamics of a Dual SARS-CoV-2 Lineage Co-Infection on a Prolonged Viral Shedding COVID-19 Case: Insights into Clinical Severity and Disease Duration. Microorganisms 2021, 9, 300. [Google Scholar] [CrossRef]
- Taghizadeh, P.; Salehi, S.; Heshmati, A.; Houshmand, S.M.; Inanloo Rahatloo, K.; Mahjoubi, F.; Sanati, M.H.; Yari, H.; Alavi, A.; Jamehdar, S.A.; et al. Study on SARS-CoV-2 strains in Iran reveals potential contribution of co-infection with and recombination between different strains to the emergence of new strains. Virology 2021, 562, 63–73. [Google Scholar] [CrossRef]
- da Silva Francisco, R., Jr.; Benites, L.F.; Lamarca, A.P.; de Almeida, L.G.P.; Hansen, A.W.; Gularte, J.S.; Demoliner, M.; Gerber, A.L.; Guimarães, A.P.d.C.; Antunes, A.K.E. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. 2021, 296, 198345. [Google Scholar] [CrossRef] [PubMed]
- Artesi, M.; Bontems, S.; Göbbels, P.; Franckh, M.; Maes, P.; Boreux, R.; Meex, C.; Melin, P.; Hayette, M.P.; Bours, V.; et al. A Recurrent Mutation at Position 26340 of SARS-CoV-2 Is Associated with Failure of the E Gene Quantitative Reverse Transcription-PCR Utilized in a Commercial Dual-Target Diagnostic Assay. J. Clin. Microbiol. 2020, 58, e01598-20. [Google Scholar] [CrossRef] [PubMed]
- Kriegova, E.; Fillerova, R.; Raska, M.; Manakova, J.; Dihel, M.; Janca, O.; Sauer, P.; Klimkova, M.; Strakova, P.; Kvapil, P. Excellent option for mass testing during the SARS-CoV-2 pandemic: Painless self-collection and direct RT-qPCR. Virol. J. 2021, 18, 95. [Google Scholar] [CrossRef] [PubMed]
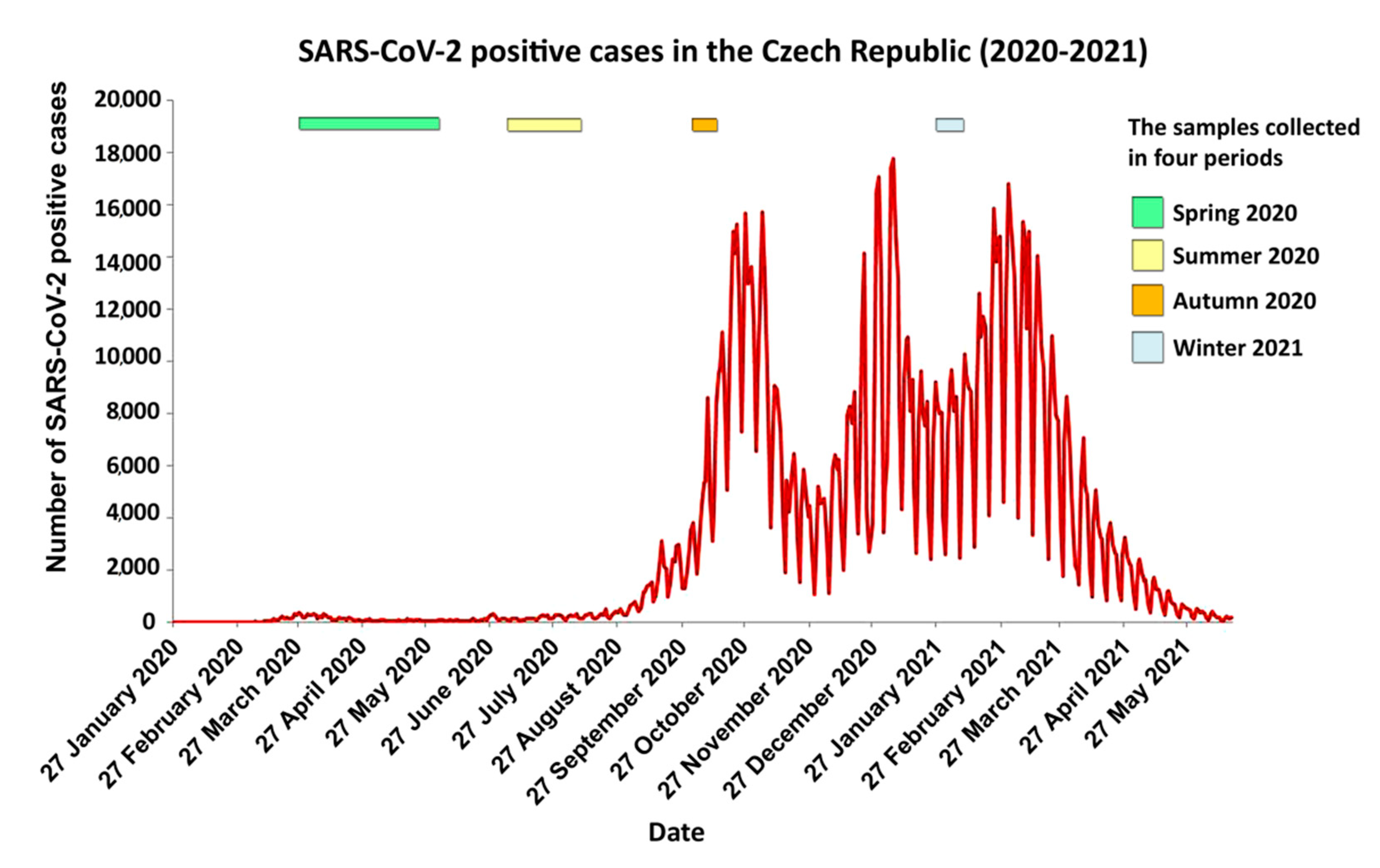

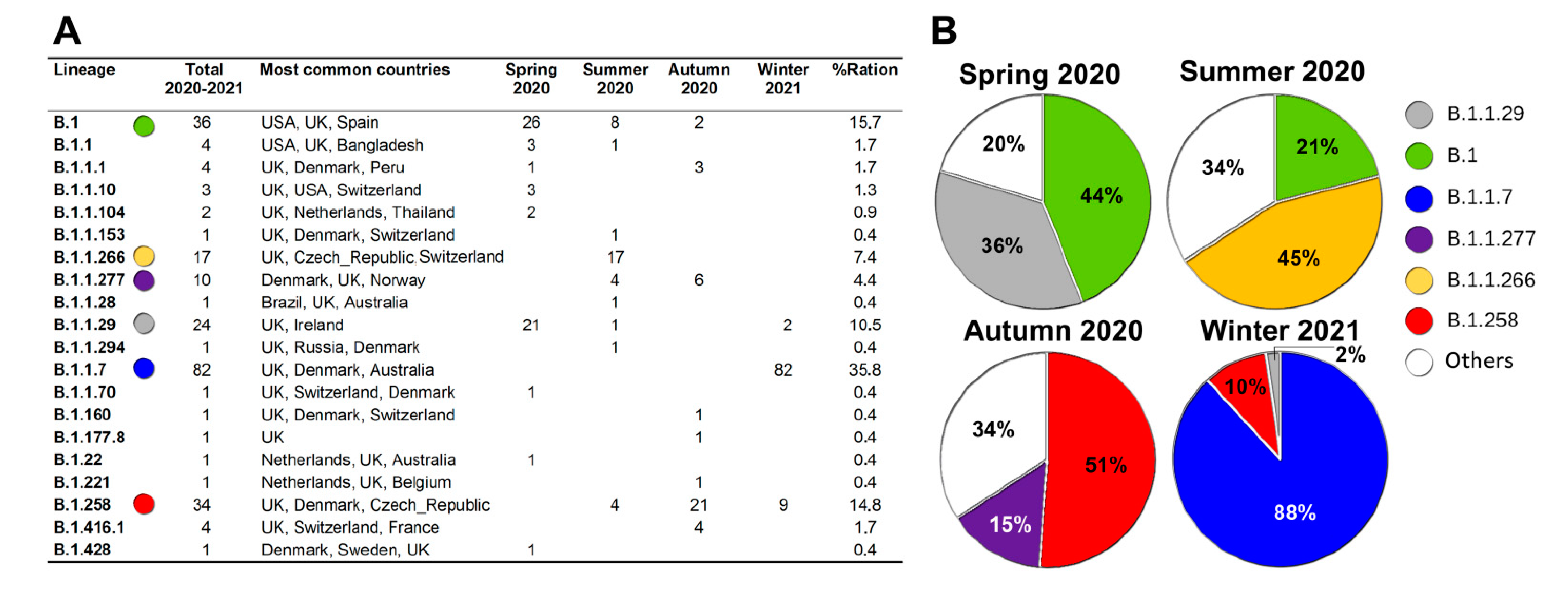
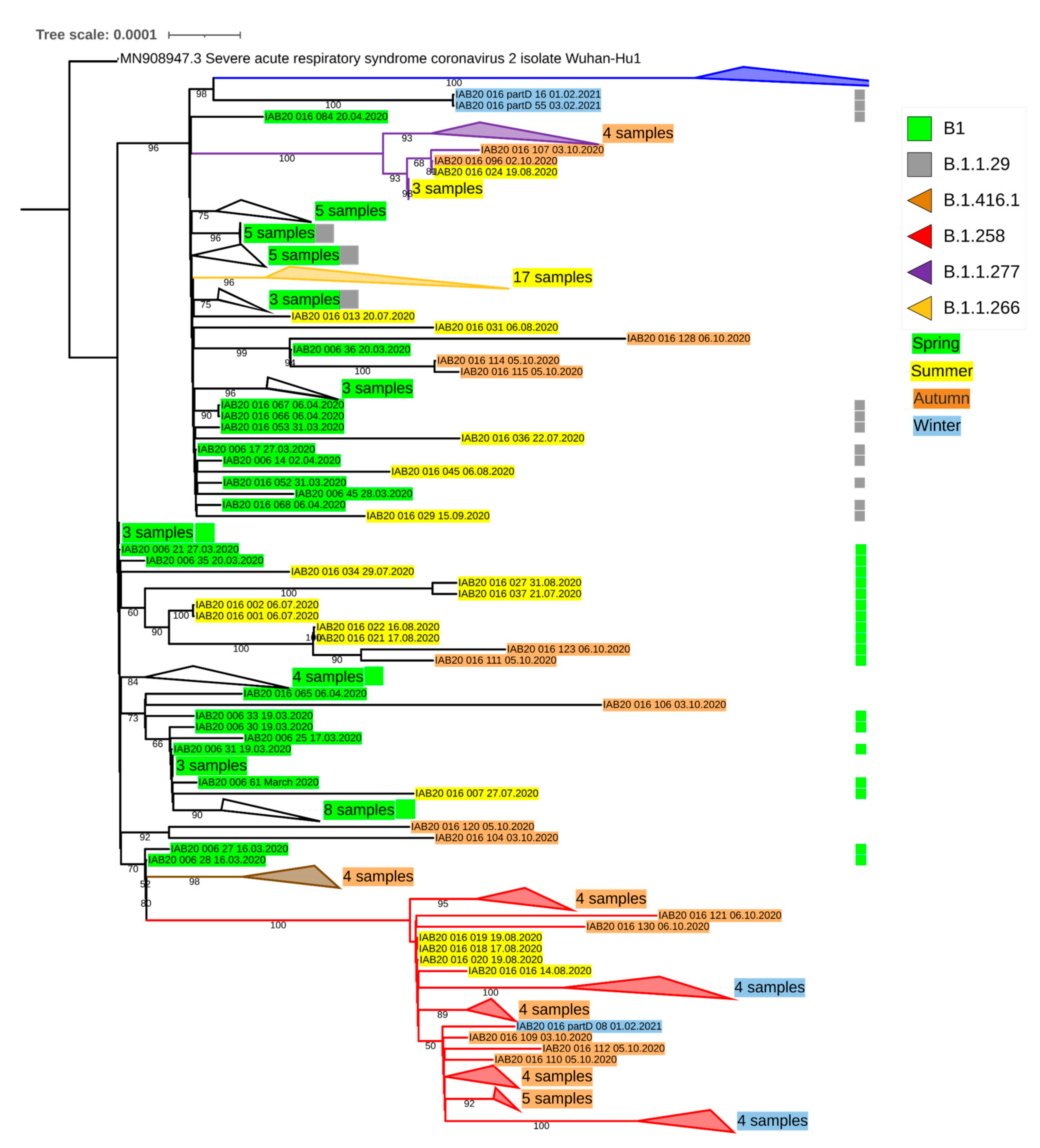

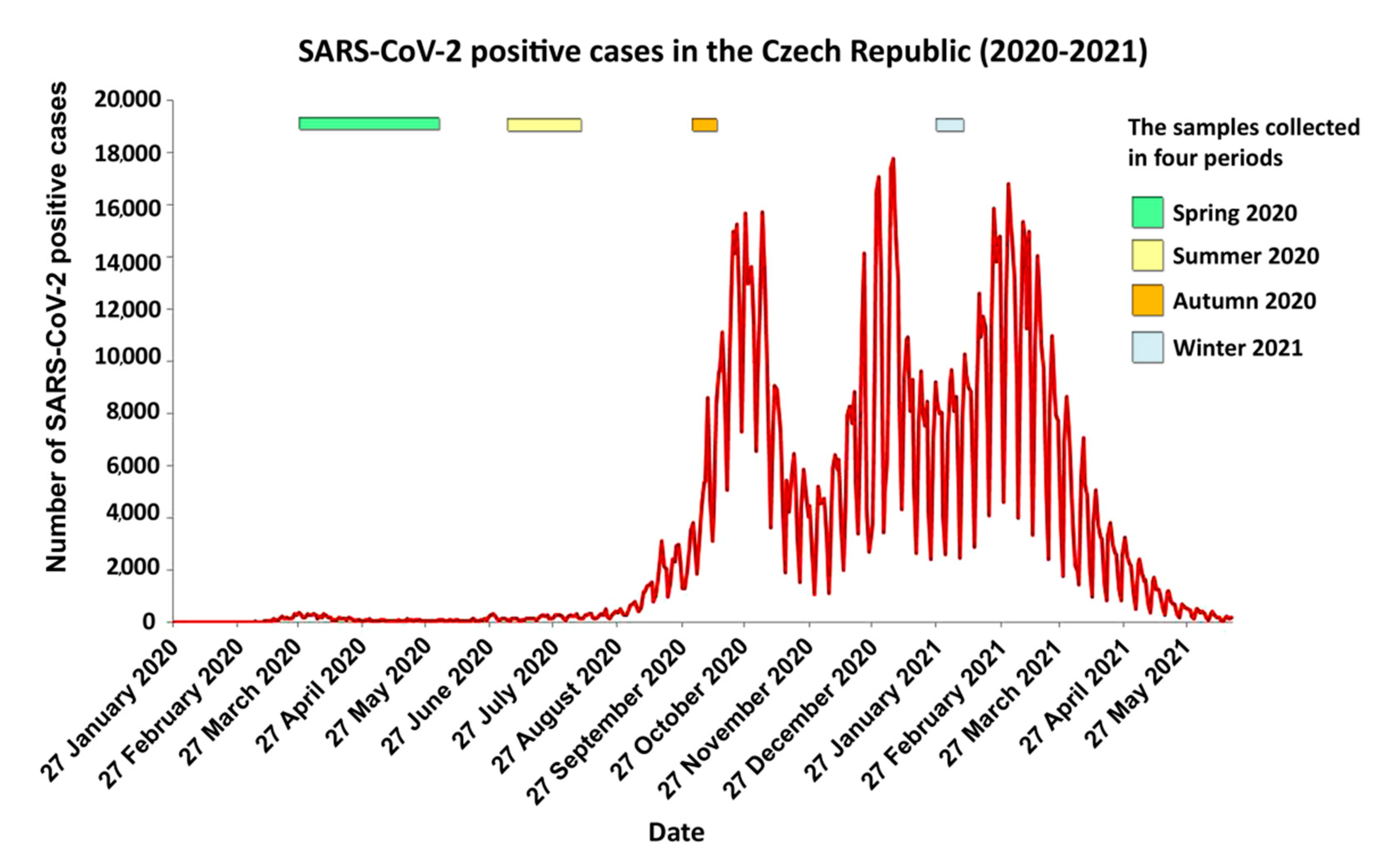{kind=link}
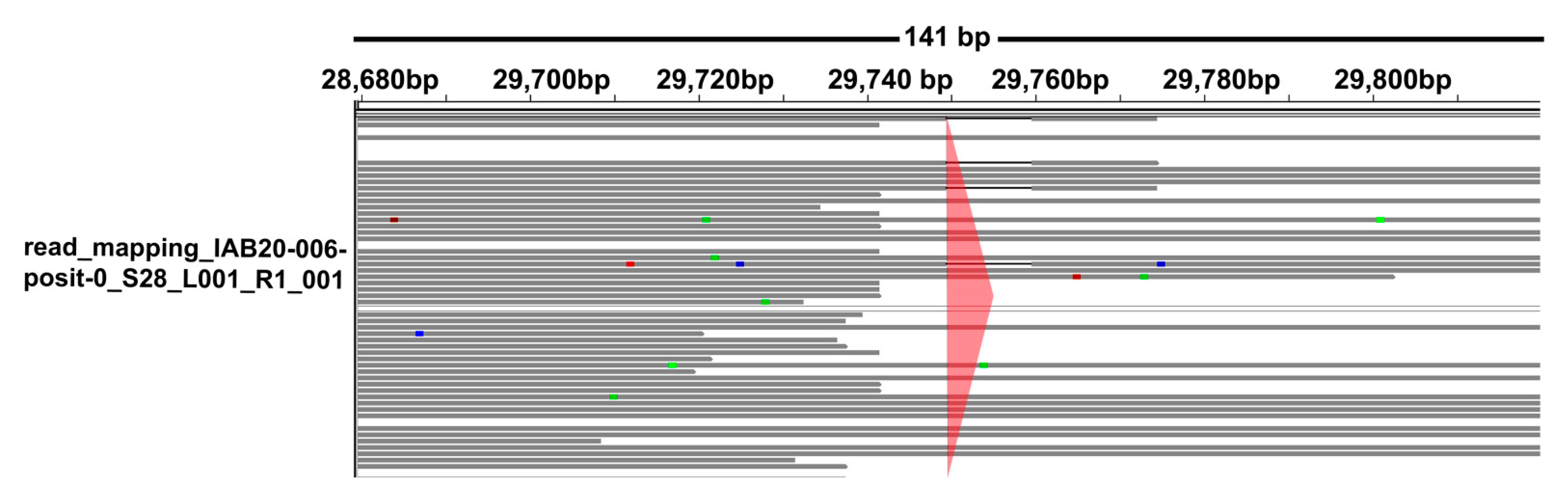{kind=link}
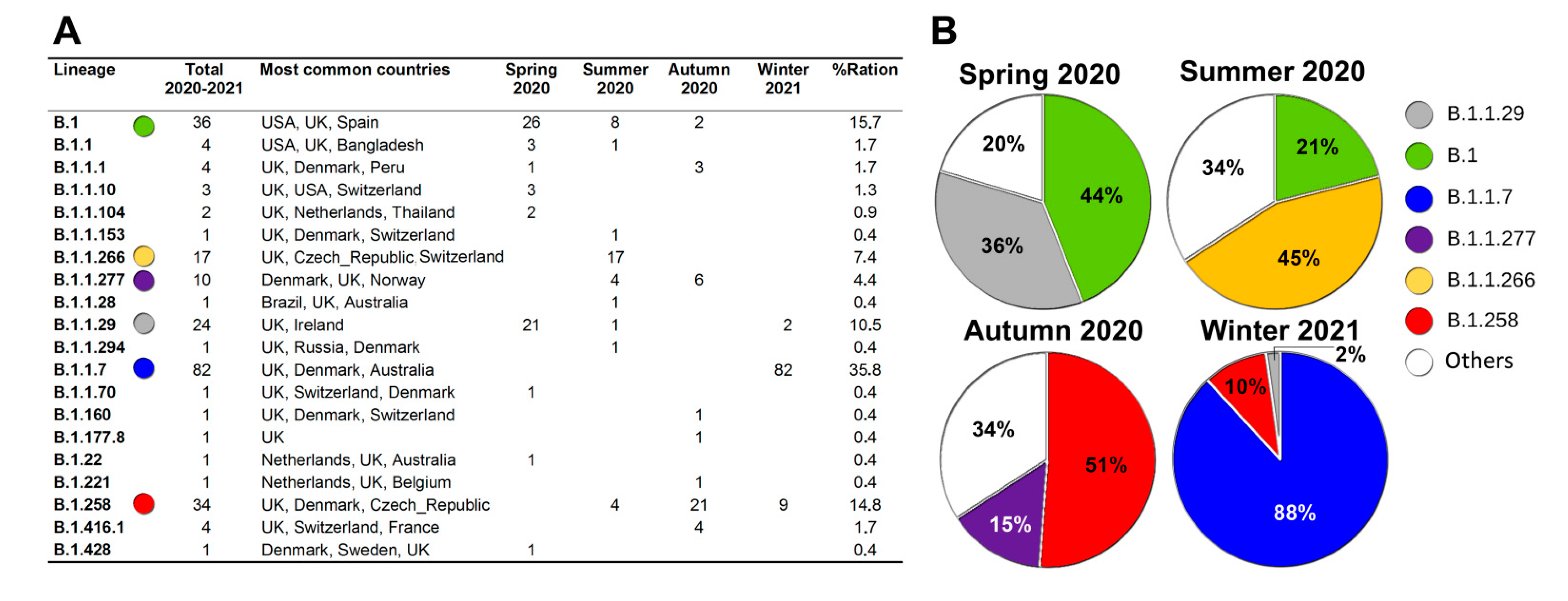{kind=link}
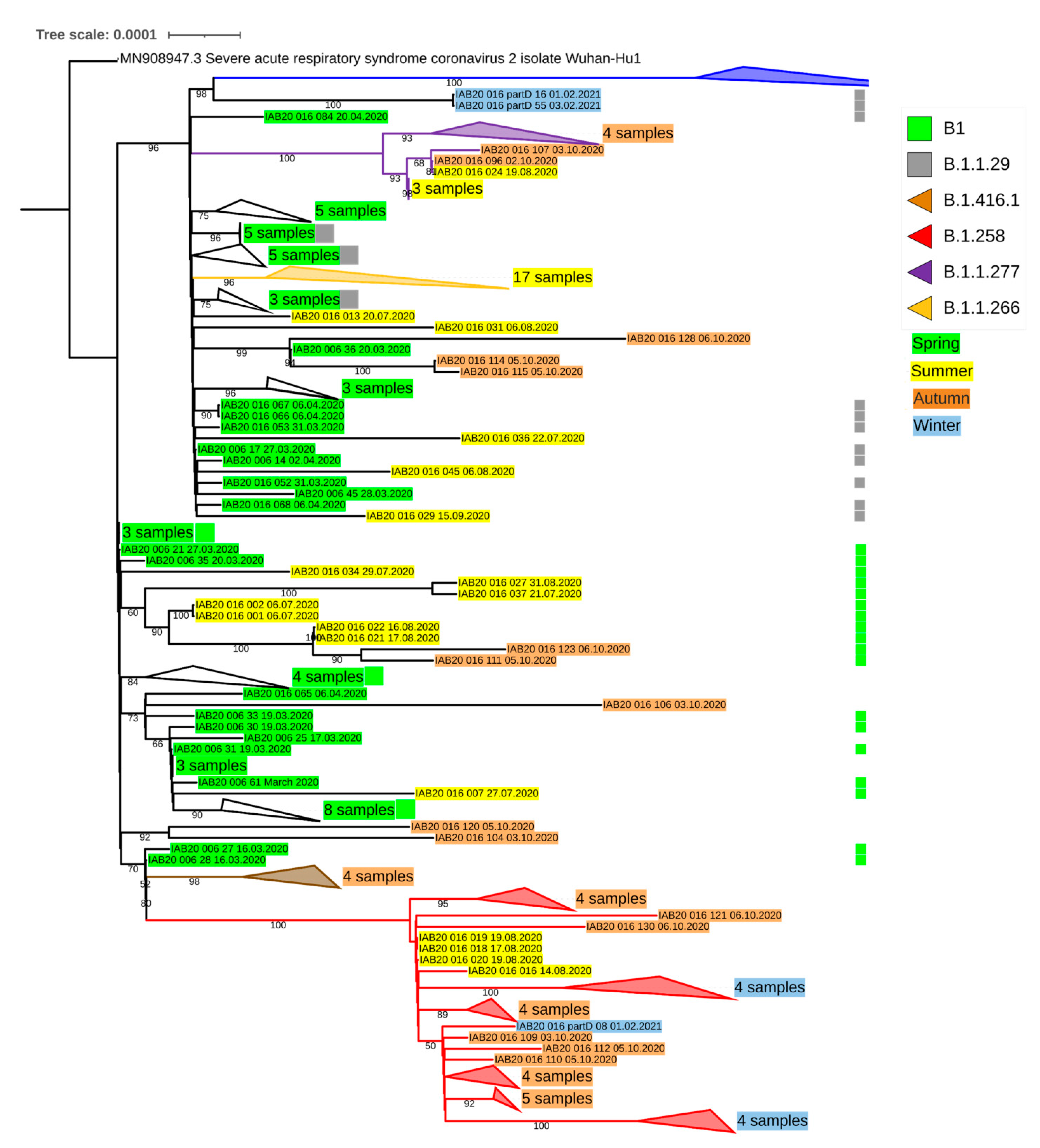{kind=link}
| Period | Month | Number of Samples | Number of Samples/Period |
|---|---|---|---|
| March | 41 | ||
| Spring 2020 | April | 15 | 59 |
| June | 3 | ||
| July | 13 | ||
| Summer 2020 | August | 24 | 38 |
| September | 1 | ||
| Autumn 2020 | October | 39 | 39 |
| Winter 2021 | January | 6 | 93 |
| February | 87 | ||
| Total | 229 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klempt, P.; Brzoň, O.; Kašný, M.; Kvapilová, K.; Hubáček, P.; Briksi, A.; Bezdíček, M.; Koudeláková, V.; Lengerová, M.; Hajdúch, M.; et al. Distribution of SARS-CoV-2 Lineages in the Czech Republic, Analysis of Data from the First Year of the Pandemic. Microorganisms 2021, 9, 1671. https://doi.org/10.3390/microorganisms9081671
Klempt P, Brzoň O, Kašný M, Kvapilová K, Hubáček P, Briksi A, Bezdíček M, Koudeláková V, Lengerová M, Hajdúch M, et al. Distribution of SARS-CoV-2 Lineages in the Czech Republic, Analysis of Data from the First Year of the Pandemic. Microorganisms. 2021; 9(8):1671. https://doi.org/10.3390/microorganisms9081671
Chicago/Turabian StyleKlempt, Petr, Ondřej Brzoň, Martin Kašný, Kateřina Kvapilová, Petr Hubáček, Aleš Briksi, Matěj Bezdíček, Vladimira Koudeláková, Martina Lengerová, Marian Hajdúch, and et al. 2021. "Distribution of SARS-CoV-2 Lineages in the Czech Republic, Analysis of Data from the First Year of the Pandemic" Microorganisms 9, no. 8: 1671. https://doi.org/10.3390/microorganisms9081671
APA StyleKlempt, P., Brzoň, O., Kašný, M., Kvapilová, K., Hubáček, P., Briksi, A., Bezdíček, M., Koudeláková, V., Lengerová, M., Hajdúch, M., Dřevínek, P., Pospíšilová, Š., Kriegová, E., Macek, M., & Kvapil, P. (2021). Distribution of SARS-CoV-2 Lineages in the Czech Republic, Analysis of Data from the First Year of the Pandemic. Microorganisms, 9(8), 1671. https://doi.org/10.3390/microorganisms9081671