
Sasso Pisano Geothermal Field Environment Harbours Diverse Ktedonobacteria Representatives and Illustrates Habitat-Specific Adaptations



Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Environmental DNA Extraction, Amplicon Generation and Sequencing
2.3. Processing of 16S rRNA Gene-Based Amplicon Sequencing Data
2.4. Analysis of 18S rRNA Gene-Based Amplicon Sequencing Data
2.5. Transmission Electron Microscopy (TEM)
2.6. Prokaryotic Community Functional Profiling
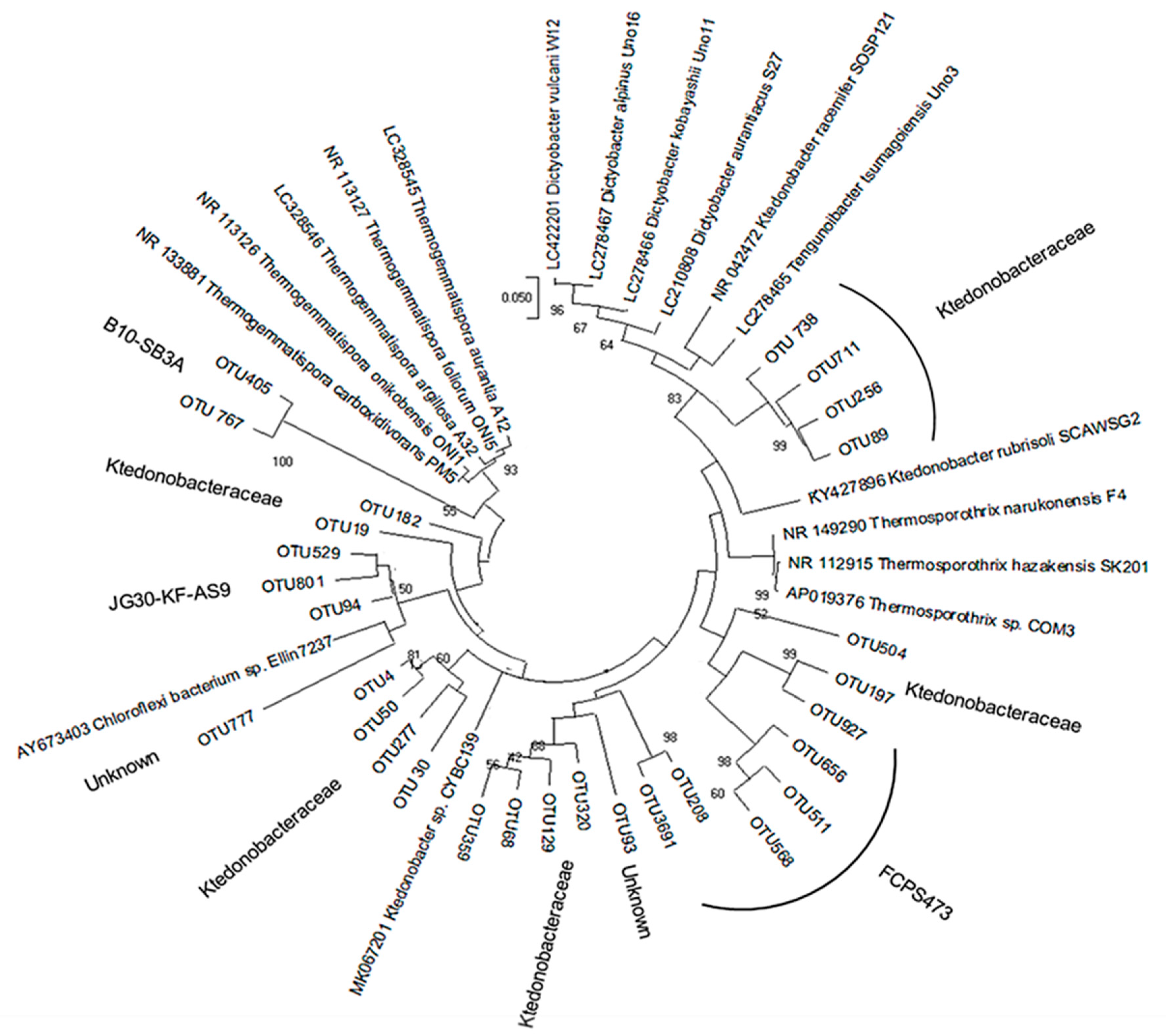
2.7. Phylogenetic Analysis
3. Results
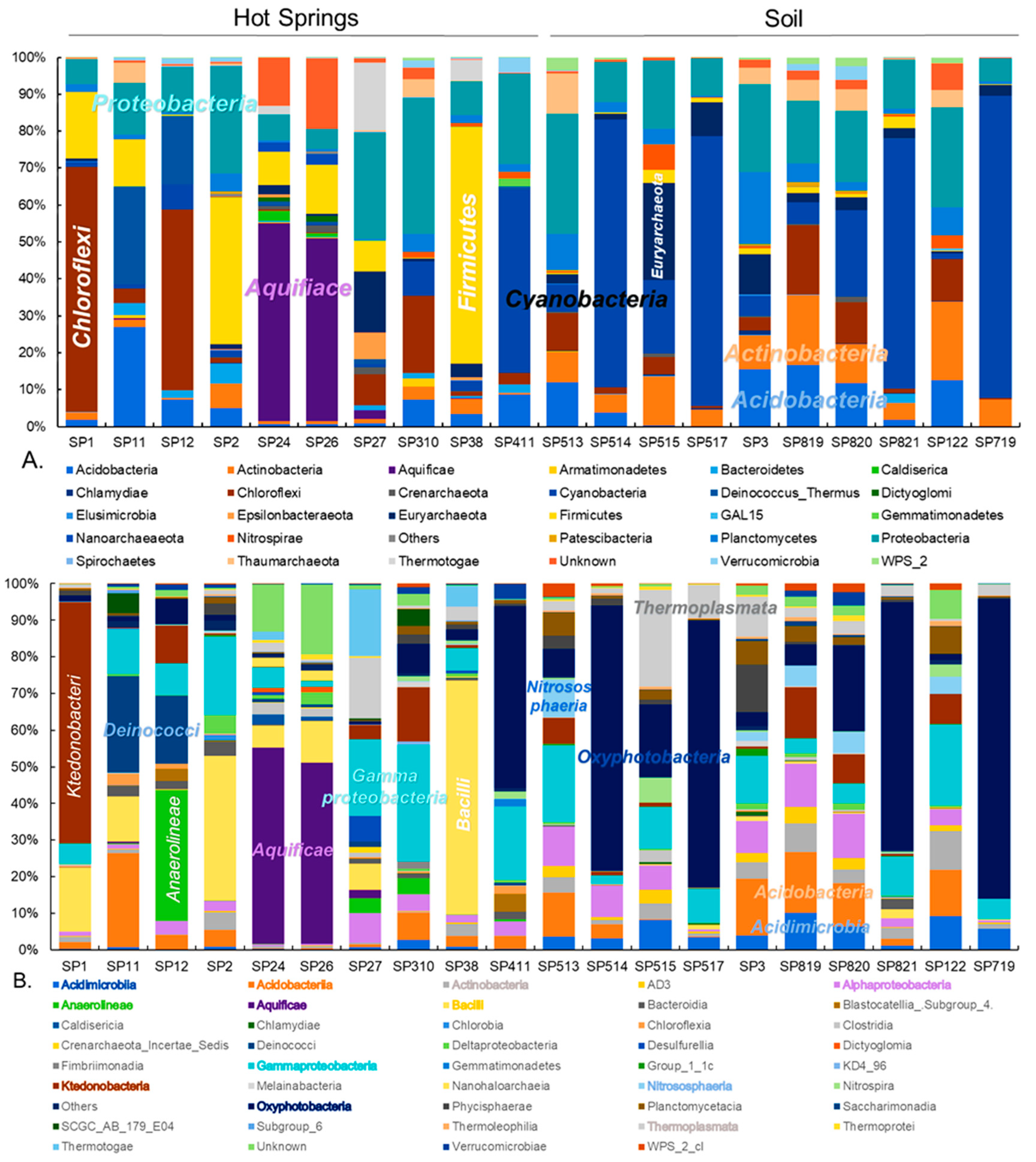
3.1. Prokaryotic Community Composition
3.2. Diversity of Ktedonobacteria
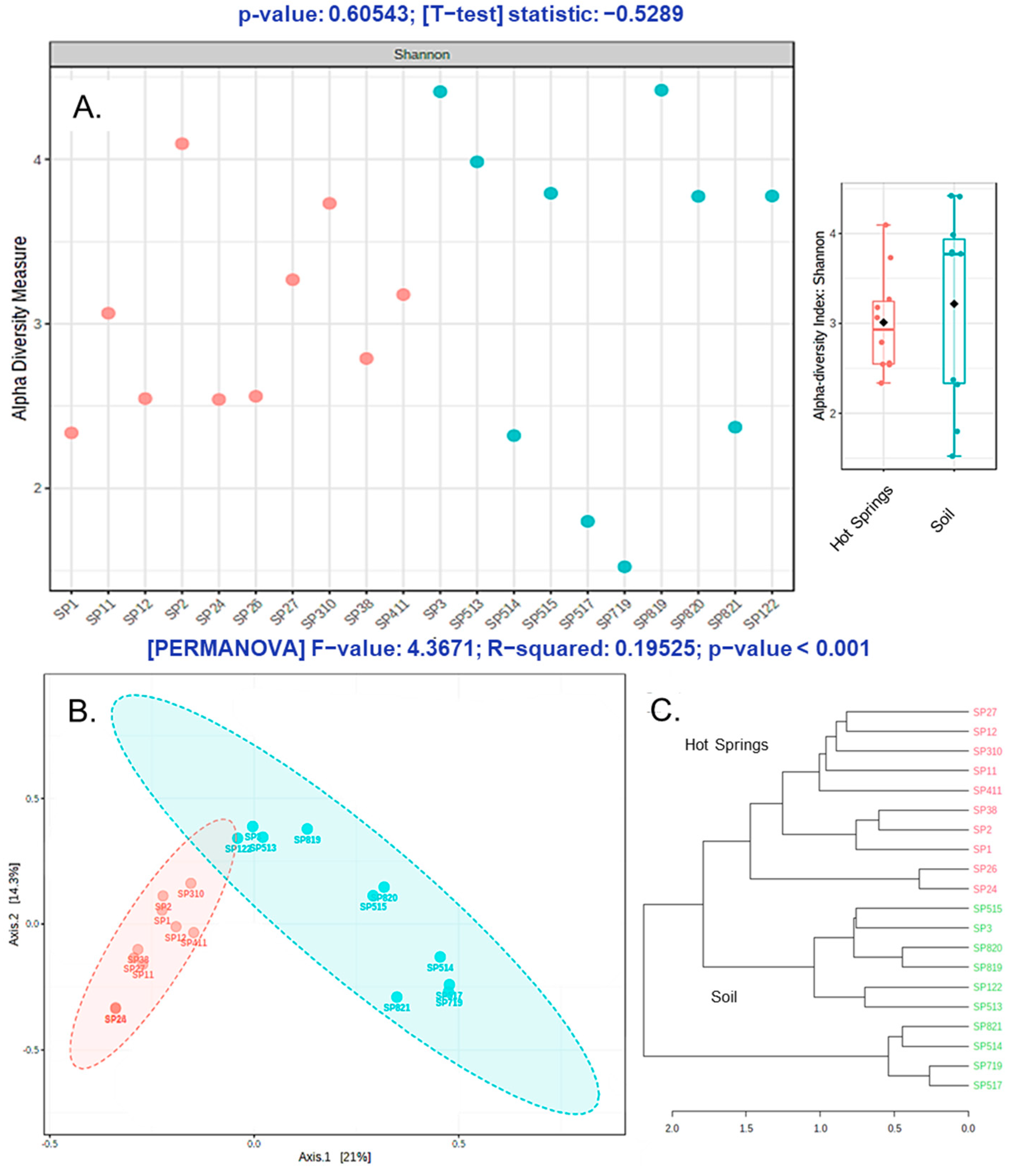
3.3. Alpha and Beta Diversity
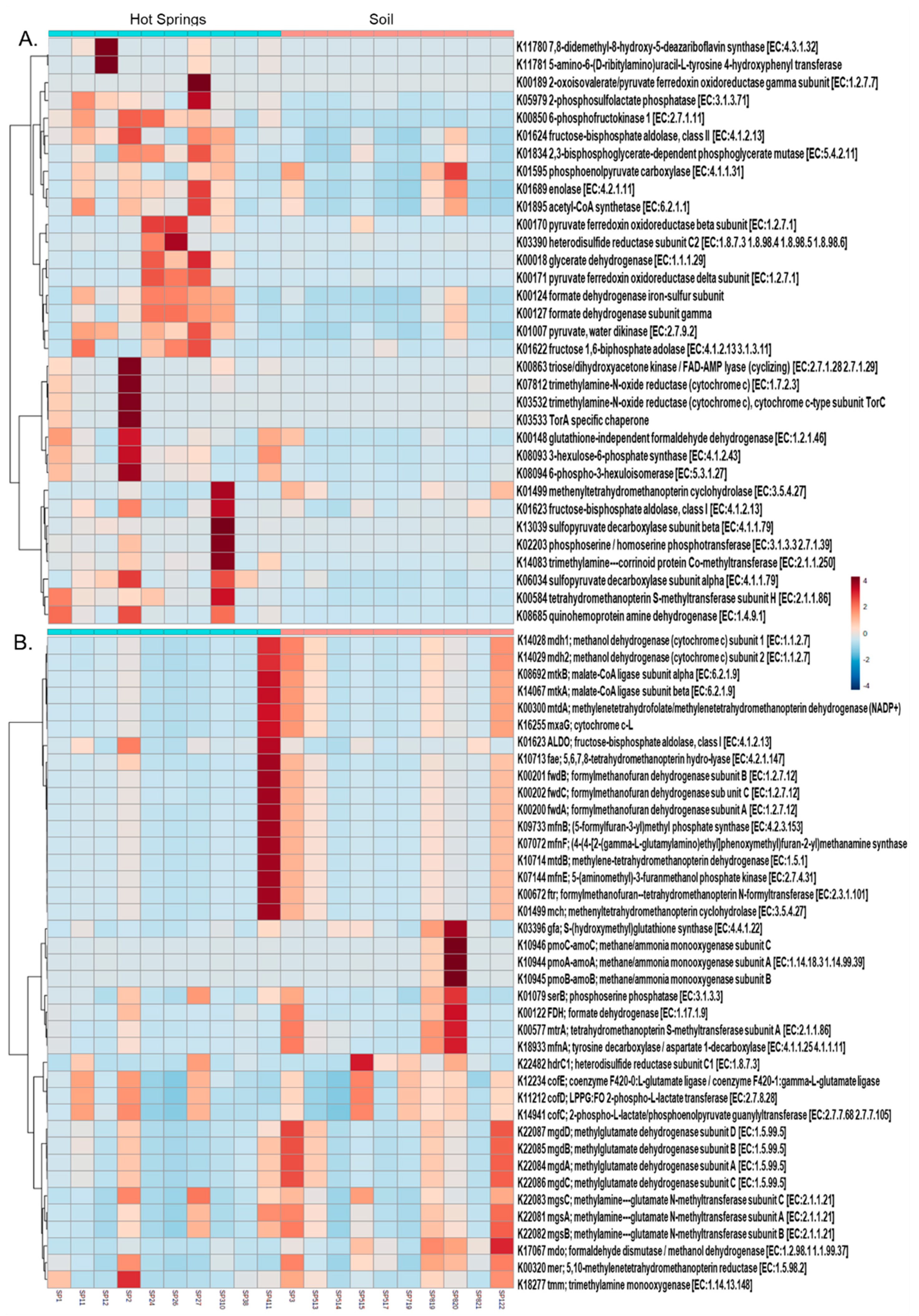
3.4. Functional Profile
3.5. Eukaryotic Diversity at a Fumarole
3.6. Structural and Morphological Description of a Eukaryotic Biofilm
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barelli, A.; Bertini, G.; Buonasorte, G.; Fiordelisi, A.; Fiordelisi, C. Recent deep exploration results at the margins of the Larderello-Travale geothermal system. In Proceedings of the World Geothermal Congress 2000, Kyushu, Japan, 28 May–10 June 2000. [Google Scholar]
- Bartoli, G.; Bottega, S.; Forino, L.M.C.; Ciccarelli, D.; Spano, C. Plant adaptation to extreme environments: The example of Cistus salviifolius of an active geo-thermal alteration field. C. R. Biol. 2014, 337, 101–110. [Google Scholar] [CrossRef]
- Bertini, G.; Casini, M.; Gianelli, G.; Pandeli, E. Geological structure of a long-living geothermal system, Larderello, Italy. Terra Nova 2006, 18, 163–169. [Google Scholar] [CrossRef]
- Narasimhan, T.; Goyal, K. Subsidence due to geothermal fluid withdrawal. Subsid. Geotherm. Fluid Withdraw. 1982, 6, 35–36. [Google Scholar] [CrossRef][Green Version]
- Bussotti, F.; Tognelli, R.; Montagni, G.; Borghini, F.; Bruschi, P.; Tani, C. Response of Quercus pubescens leaves exposed to geothermal pollutant input in southern Tuscany (Italy). Environ. Pollut. 2003, 121, 349–361. [Google Scholar] [CrossRef]
- Pippucci, A.; Lorenzi, R.; Spanò, C.; Sorce, C. Stress-induced changes to the flora in a geothermal field in central Italy. Acta Physiol. Plant 2015, 37, 1–10. [Google Scholar] [CrossRef]
- Brogi, A. The structure of the Monte Amiata volcano-geothermal area (Northern Apennines, Italy): Neogene-Quaternary compression versus extension. Intl. J. Earth Sci. 2007, 97, 677–703. [Google Scholar] [CrossRef]
- Zelenski, M.; Taran, Y.; Galle, B. High emission rate of sulfuric acid from Bezymianny volcano, Kamchatka. Geophys. Res. Lett. 2015, 42, 7005–7013. [Google Scholar] [CrossRef]
- D’Amore, F.; Panichi, C. Evaluation of deep temperatures of hydrothermal systems by a new gas geothermometer. Geochim. Cosmochim. Acta 1980, 44, 549–556. [Google Scholar] [CrossRef]
- Selvi, F. Geothermal Biotopes in Central Western Italy from a Botanical Viewpoint. Ecosystem Responses to CO2: The MAPLE Project Results; Official Publications of the European Communities; European Communities: Luxemburg, 1999. [Google Scholar]
- Elmarsdottir, A.; Ingimarsdottir, M.; Hansen, I.; Olafsson, J.S.; Olafsson, E. Vegetation and invertebrates in three geothermal areas in Iceland. In Proceedings of the International Geothermal Conference, Reykjavik, Iceland, 14–17 September 2003; Citeseer: Reykjavik, Iceland, 2003. [Google Scholar]
- Rothschild, L.J.; Mancinelli, R.L. Life in extreme environments. Nat. Cell Biol. 2001, 409, 1092–1101. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Stoeck, T.; Bass, D.; Nebel, M.; Christen, R.; Jones, M.D.M.; Breiner, H.-W.; Richards, T.A. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 2010, 19, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.; Schneider, D.; Hoppert, M. Bioleaching potential of bacterial communities in historic mine waste areas. Environ. Earth Sci. 2018, 77, 542. [Google Scholar] [CrossRef]
- Dong, X.; Kleiner, M.; Sharp, C.E.; Thorson, E.; Li, C.; Liu, D.; Strous, M. Fast and simple analysis of MiSeq amplicon sequencing data with MetaAmp. Front. Microbiol. 2017, 8, 1461. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformation 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A fast, accurate, splice-aware aligner. In Proceedings of the 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, USA, 20 March 2014; Lawrence Berkeley National Lab: Walnut Creek, CA, USA, 2014. [Google Scholar]
- Rognes, T.; Flouri, T.; Nicholas, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmas, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The Silva ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G.I. Mikrobiome Helper: A custom and streamlined workflow for microbiome research. Meth. Protoc. 2017, 2, 11. [Google Scholar]
- Iwai, S.; Weinmaier, T.; Schmidt, B.L.; Albertson, D.G.; Poloso, N.J.; Dabbagh, K.; DeSantis, T.Z. Piphillin: Improved prediction of metagenomic content by direct inference from human microbiomes. PLoS ONE 2016, 11, e0166104. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Nazem-Bokaee, H.; Gopalakrishnan, S.; Ferry, J.G.; Wood, T.K.; Maranas, C.D. Assessing methanotrophy and carbon fixation for biofuel production by Methanosarcina acetivorans. Microb. Cell Factor. 2016, 15, 1–13. [Google Scholar] [CrossRef]
- Bennett, R.K.; Steinberg, L.M.; Chen, W.; Papoutsakis, E.T. Engineering the bioconversion of methane and methanol to fuels and chemicals in native and synthetic methylotrophs. Curr. Opin. Biotechnol. 2018, 50, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.Y.; Nguyen, A.D.; Nguyen, T.T.; Nguyen, L.T.; Lee, O.K.; Lee, E.Y. Biological conversion of methane to chemicals and fuels: Technical challenges and issues. Appl. Microbiol. Biotechnol. 2018, 102, 3071–3080. [Google Scholar] [CrossRef]
- Sømme, L. Invertebrates in Hot and Cold Arid Environments; Springer: Berlin/Heidelberg, Germany, 1995; p. 275. [Google Scholar]
- Ciniglia, C.; Yoon, H.S.; Pollio, A.; Pinto, G.; Bhattacharya, D. Hidden biodiversity of the extremophilic Cyanidiales red algae. Mol. Ecol. 2004, 13, 1827–1838. [Google Scholar] [CrossRef]
- Zheng, Y.; Saitou, A.; Wang, C.-M.; Toyoda, A.; Minakuchi, Y.; Sekiguchi, Y.; Ueda, K.; Takano, H.; Sakai, Y.; Abe, K.; et al. Genome Features and Secondary Metabolites Biosynthetic Potential of the Class Ktedonobacteria. Front. Microbiol. 2019, 10, 893. [Google Scholar] [CrossRef] [PubMed]
- Yabe, S.; Aiba, Y.; Sakai, Y.; Hazaka, M.; Yokota, A. Thermosporothrix hazakensis gen. nov., sp. nov., isolated from compost, description of Thermospo-rotrichaceae fam. nov. within the class Ktedonobacteria Cavaletti et al. 2007 and emended description of the class Ktedonobacteria. Int. J. Syst. Evol. Microbiol. 2010, 60, 1794–1801. [Google Scholar] [CrossRef]
- Yabe, S.; Aiba, Y.; Sakai, Y.; Hazaka, M.; Yokota, A. Thermogemmatispora onikobensis gen. nov., sp. nov. and Thermogemmatispora foliorum sp. nov., isolated from fallen leaves on geothermal soils, and description of Thermogemmatisporaceae fam. nov. and Thermogemmatisporales ord. nov. within the class Ktedonobacteria. Int. J. Syst. Evol. Microbiol. 2011, 61, 903–910. [Google Scholar]
- Cavaletti, L.; Monciardini, P.; Bamonte, R.; Schumann, P.; Rohde, M.; Sosio, M.; Donadio, S. New Lineage of filamentous, spore-forming, Gram-positive bacteria from soil. Appl. Environ. Microbiol. 2006, 72, 4360–4369. [Google Scholar] [CrossRef] [PubMed]
- Yabe, S.; Sakai, Y.; Abe, K.; Yokota, A.; Také, A.; Matsumoto, A.; Sugiharto, A.; Susilowati, D.; Hamada, M.; Nara, K.; et al. Dictyobacter aurantiacus gen. nov., sp. nov., a member of the family Ktedonobacteraceae, isolated from soil, and emended description of the genus Thermosporothrix. Int. J. Syst. Evol. Microbiol. 2017, 67, 2615–2621. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, P.; Jiang, D.; Dai, X.; Zhang, R.; Wang, Y.; Wang, Y. Microbial community structure and arsenic biogeochemistry in an acid vapor-formed spring in Tengchong Geothermal Area, China. PLoS ONE 2016, 11, e0146331. [Google Scholar] [CrossRef]
- De Miera, L.E.S.; Arroyo, P.; de Luis Calabuig, E.; Falagan, J.; Ansola, G. High-throughput sequencing of 16S RNA genes of soil bacterial communities from a naturally occurring CO2 gas vent. Int. J. Greenh. Gas. Control. 2014, 29, 176–184. [Google Scholar] [CrossRef]
- Northup, D.; Melim, L.; Spilde, M.; Hathaway, J.; Garcia, M.; Moya, M.; Stone, F.; Boston, P.; Dapkevicius, M.; Riquelme, C. Lava cave microbial communities within mats and secondary mineral deposits: Implications for life detection on other planets. Astrobiology 2011, 11, 601–618. [Google Scholar] [CrossRef]
- Brito, E.M.S.; Romero-Núñez, V.M.; Caretta, C.A.; Bertin, P.; Valerdi-Negreros, J.C.; Guyoneaud, R.; Goñi-Urriza, M. The bacterial diversity on steam vents from Paricutín and Sapichu volcanoes. Extremophiles 2019, 23, 249–263. [Google Scholar] [CrossRef]
- Yabe, S.; Sakai, Y.; Abe, K.; Yokota, A. Diversity of Ktedonobacteria with Actinomycetes-like morphology in terrestrial environments. Microbes Environ. 2017, 32, 61–70. [Google Scholar] [CrossRef]
- Ghezzi, D.; Sauro, F.; Columbu, A.; Carbone, C.; Hong, P.-Y.; Vergara, F.; De Waele, J.; Cappelletti, M. Transition from unclassified Ktedonobacterales to Actinobacteria during amorphous silica precipitation in a quartzite cave environment. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- King, C.E.; King, G.M. Description of Thermogemmatispora carboxidivorans sp. nov., a carbon-monoxide-oxidizing member of the class Ktedonobacteria isolated from a geothermally heated biofilm, and analysis of carbon monoxide oxidation by members of the class Ktedonobacteria. Int. J. Syst. Evol. Microbiol. 2014, 64, 1244–1251. [Google Scholar] [CrossRef]
- Arif, S.; Nacke, H.; Hoppert, M. Metagenome-assembled genome sequences of a biofilm derived from Marsberg Copper Mine. Microbiol. Res. Announ. 2021, 10, e01253-20. [Google Scholar]
- Wall, K.; Cornell, J.; Bizzoco, R.W.; Kelley, S.T. Biodiversity hot spot on a hot spot: Novel extremophile diversity in Hawaiian fumaroles. Microbiology 2015, 4, 267–281. [Google Scholar] [CrossRef]
- Tassi, F.; Venturi, S.; Cabassi, J.; Capecchiacci, F.; Nisi, B.; Vaselli, O. Volatile organic compounds (VOCs) in soil gases from Solfatara crater (Campi Flegrei, southern Italy): Geogenic source(s) vs. biogeochemical processes. Appl. Geochem. 2015, 56, 37–49. [Google Scholar] [CrossRef]
- Ji, M.; Greening, C.; Vanwonterghem, I.; Carere, C.R.; Bay, S.K.; Steen, J.A.; Montgomery, K.; Lines, T.; Beardall, J.; Van Dorst, J.; et al. Atmospheric trace gases support primary production in Antarctic desert surface soil. Nat. Cell Biol. 2017, 552, 400–403. [Google Scholar] [CrossRef] [PubMed]
- King, G.M.; Weber, C.F.; Nanba, K.; Sato, Y.; Ohta, H. Atmospheric CO and hydrogen uptake and CO oxidizer phylogeny for Miyakejima, Japan volcanic deposits. Microb. Environ. 2008, 23, 229. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, E.; Jiang, H.; Li, W.; Wu, G.; Huang, L.; Hedlund, B.P.; Dong, H. Distribution and diversity of aerobic carbon monoxide-oxidizing bacteria in geothermal springs of China, the Philippines, and the United States. Geomicrobiol. J. 2015, 32, 903–913. [Google Scholar] [CrossRef]
- Arce-Rodríguez, A.; Puente-Sánchez, F.; Avendaño, R.; Martínez-Cruz, M.; De Moor, J.M.; Pieper, D.H.; Chavarría, M. Thermoplasmatales and sulfur-oxidizing bacteria dominate the microbial community at the surface water of a CO2-rich hydrothermal spring located in Tenorio Volcano National Park, Costa Rica. Extremophiles 2019, 23, 177–187. [Google Scholar] [CrossRef] [PubMed]
- De Miera, L.E.S.; Arroyo, P.; de Luis Calabuig, E.; Ansola, G. Effects of varying CO2 flows on bacterial communities in mesocosms created from two soils. Int. J. Greenh. Gas. Control. 2016, 46, 205–214. [Google Scholar] [CrossRef]
- McIlroy, S.J.; Kirkegaard, R.H.; Dueholm, M.S.; Fernando, E.; Karst, S.M.; Albertsen, M.; Nielsen, P.H. Culture-independent analyses reveal novel Anaerolineaceae as abundant primary fermenters in anaerobic digesters treating waste activated sludge. Front. Microbiol. 2017, 8, 1134. [Google Scholar] [CrossRef] [PubMed]
- Kochetkova, T.V.; Toshchakov, S.V.; Zayulina, K.S.; Elcheninov, A.G.; Zavarzina, D.G.; Lavrushin, V.Y.; Bonch-Osmolovskaya, E.A.; Kublanov, I.V. Hot in Cold: Microbial life in the hottest springs in permafrost. Microorganisms 2020, 8, 1308. [Google Scholar] [CrossRef]
- Nishihara, A.; Matsuura, K.; Tank, M.; McGlynn, S.E.; Thiel, V.; Haruta, S. Nitrogenase activity in thermophilic chemolithoautotrophic bacteria in the phylum Aquificae isolated under nitrogen-fixing conditions from Nakabusa Hot Springs. Microb. Environ. 2018, 33, 394–401. [Google Scholar] [CrossRef]
- Rainey, F.A.; Nobre, M.F.; Schumann, P.; Stackebrandt, E.; Da Costa, M.S. Phylogenetic diversity of the Deinococci as determined by 16S ribosomal DNA sequence comparison. Int. J. System. Evol. Microbiol. 1997, 47, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Gerber, E.; Bernard, R.; Castang, S.; Chabot, N.; Coze, F.; Dreux-Zigha, A.; Hauser, E.; Hivin, P.; Joseph, P.; Lazarelli, C.; et al. Deinococcus as new chassis for industrial biotechnology: Biology, physiology and tools. J. Appl. Microbiol. 2015, 119, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sumner, D.; Hawes, I.; Mackey, T.J.; Jungblut, A.D.; Doran, P.T. Antarctic microbial mats: A modern analog for Archean lacustrine oxygen oases. Geology 2015, 43, 887–890. [Google Scholar] [CrossRef]
- Hamilton, T.L.; Klatt, J.M.; de Beer, D.; Macalady, J.L. Cyanobacterial photosynthesis under sulfidic conditions: Insights from the isolate Leptolyngbya sp. strain hensonii. ISME J. 2018, 12, 568–584. [Google Scholar] [CrossRef]
- De Luca, P.; Taddei, R.; Varano, L. Cyanidioschyzom merolae: A new alga of thermal acidic enviroments. J. Plant Taxon. Geogr. 1978, 33. [Google Scholar]
- Merola, A.; Castaldo, R.; De Luca, P.; Gambardella, R.; Musacchio, A.; Taddei, R. Revision of Cyanidium caldarium. Three species of acidophilic algae. G. Bot. Ital. 1981, 115, 189–195. [Google Scholar] [CrossRef]
- Yoon, H.S.; Ciniglia, C.; Wu, M.; Comeron, J.M.; Pinto, G.; Pollio, A.; Bhattacharya, D. Establishment of endolithic populations of extremophilic Cyanidiales (Rhodophyta). BMC Evol. Biol. 2006, 6, 78. [Google Scholar] [CrossRef][Green Version]
- Kühn, J.; Ruess, L. Effects of resource quality on the fitness of collembola fed single and mixed diets from the green and brown food chain. Soil Biol. Biochem. 2021, 154, 108156. [Google Scholar] [CrossRef]
- Scheu, S.; Folger, M. Single and mixed diets in Collembola: Effects on reproduction and stable isotope fractionation. Funct. Ecol. 2004, 18, 94–102. [Google Scholar] [CrossRef]
- Haubert, D.; Häggblom, M.M.; Langel, R.; Scheu, S.; Ruess, L. Trophic shift of stable isotopes and fatty acids in Collembola on bacterial diets. Soil Biol. Biochem. 2006, 38, 2004–2007. [Google Scholar] [CrossRef]
- Mehlhorn, H. Encyclopedia of Parasitology, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transport System | |
|---|---|
| Manganese | Zinc |
| K19973 mntA; manganese ATP-binding protein K19975 mntC; manganese substrate-binding protein K19976 mntB; manganese permease protein K11601 mntC; manganese substrate-binding protein K11603 mntA; manganese ATP-binding protein K11602 mntB; manganese permease protein | K11707 troA; manganese/zinc/iron substrate-binding protein K11708 troC; manganese/zinc/iron permease protein K11709 troD; manganese/zinc/iron permease protein K11710 troB; manganese/zinc/iron ATP- binding protein |
| Iron | Copper |
| K11604 sitA; manganese/iron substrate-binding protein K11605 sitC; manganese/iron permease protein K11606 sitD; manganese/iron permease protein K11607 sitB; manganese/iron ATP-binding protein K02010 afuC; iron(III) ATP-binding protein K02011 afuB; iron(III) permease protein K02012 afuA; iron(III) substrate-binding protein | K19340 nosF; Cu-processing system ATP-binding protein K19341 nosY; Cu-processing system permease protein |
| Tungstate | |
| K05772 tupA; tungstate substrate-binding protein K05773 tupB; tungstate permease protein | |
| Molybdate | Nickel |
| K02017 modC; molybdate ATP-binding protein K02018 modB; molybdate permease protein K02020 modA; molybdate substrate-binding protein | K15584 nikA; nickel substrate-binding protein K15585 nikB; nickel permease protein K15586 nikC; nickel permease protein K15587 nikD; nickel ATP-binding protein |
| Sulphate/Thiosulphate | Cobalt |
| K02045 cysA; sulfate/thiosulfate ATP-binding protein K02046 cysU; sulfate/thiosulfate permease protein K02047 cysW; sulfate/thiosulfate permease protein K02048 cysP; sulfate/thiosulfate substrate-binding protein | K02006 cbiO; cobalt/nickel ATP-binding protein K02007 cbiM; cobalt/nickel permease protein K02008 cbiQ; cobalt/nickel permease protein K02009 cbiN; cobalt/nickel transport protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arif, S.; Willenberg, C.; Dreyer, A.; Nacke, H.; Hoppert, M. Sasso Pisano Geothermal Field Environment Harbours Diverse Ktedonobacteria Representatives and Illustrates Habitat-Specific Adaptations. Microorganisms 2021, 9, 1402. https://doi.org/10.3390/microorganisms9071402
Arif S, Willenberg C, Dreyer A, Nacke H, Hoppert M. Sasso Pisano Geothermal Field Environment Harbours Diverse Ktedonobacteria Representatives and Illustrates Habitat-Specific Adaptations. Microorganisms. 2021; 9(7):1402. https://doi.org/10.3390/microorganisms9071402
Chicago/Turabian StyleArif, Sania, Corinna Willenberg, Annika Dreyer, Heiko Nacke, and Michael Hoppert. 2021. "Sasso Pisano Geothermal Field Environment Harbours Diverse Ktedonobacteria Representatives and Illustrates Habitat-Specific Adaptations" Microorganisms 9, no. 7: 1402. https://doi.org/10.3390/microorganisms9071402
APA StyleArif, S., Willenberg, C., Dreyer, A., Nacke, H., & Hoppert, M. (2021). Sasso Pisano Geothermal Field Environment Harbours Diverse Ktedonobacteria Representatives and Illustrates Habitat-Specific Adaptations. Microorganisms, 9(7), 1402. https://doi.org/10.3390/microorganisms9071402