3.1. Recovery of Metagenome-Assembled Genomes (MAGs)
Characteristic glycosidase profiles of several
Bifidobacterium species present in the gastrointestinal tract of both healthy infants and adults were investigated through a survey of MAGs recovered from gut metagenomic datasets. To this end, shotgun metagenomic data have been collected from the main sequence repositories [
16,
17]. Sequences were assembled into contigs and MAGs, and annotation of glycosidase functional domains and families codified in individual genomes was performed using the CAZy database [
23]. A total of 1806 MAGs representatives of 177 species and strains were recovered from 487 metagenomes, comprising 1339 MAGs from 447 metagenomes from 60 infants, as well as 467 MAGs from 40 metagenomes from 39 adults (
Supplementary Materials Table S1).
Escherichia coli (n = 140),
Faecalibacterium prausnitzii (n = 116), and
Ruminococcus gnavus (n = 114) showed the highest overall number of MAGs recovered. Asnicar et al. [
7] and Pasolli et al. [
8] recovered MAGs from
F. prausnitzii and
R. gnavus showing the relevance of these species in large cohorts of participants.
Escherichia,
Faecalibacterium and
Ruminococcus were also found in infant metagenomes using assembly-free methods [
16].
In general, bifidobacterial species were among the most frequently identified MAGs species, including
B. longum (n = 70),
B. bifidum (n = 39),
B. breve (n = 38) and
B. pseudocatenulatum (n = 35). However, a low number of MAGs were recovered from other bifidobacteria species, such as
B. adolescentis (n = 17),
B. dentium (n = 2),
B. catenulatum (n = 1) and
B. scardovii (n = 1). Previous studies also reported MAG recovery from
B. adolescentis,
B. bifidum,
B. breve,
B. dentium and
B. longum in fecal metagenomes from human donors following westernized and non-westernized lifestyles [
7,
8], highlighting the relevance of these species in metagenomes from different individuals (infants and adults) exposed to specific types of diets, including variable prebiotic consumption. Moreover, gene families and metabolic pathways of several species belonging to Bifidobacteriaceae were reported by previous authors in the assembly free analysis of the infant metagenomes [
16], in agreement with our assembly results.
Other species and strains showing a high number of MAGs recovered were
Collinsella aerofaciens (n = 71),
Veillonella parvula (n = 62),
Akkermansia muciniphila (n = 59), Lachnospiraceae
bacterium strain GAM79 (n = 50) and
Ruthenibacterium lactatiformans (n = 46). MAGs from
A. muciniphila were also recovered from Pasolli et al. [
8] in westernized and non-westernized cohorts in agreement with our results. It has been reported that metabolic pathways of
Collinsella,
Veillonella parvula and
Akkermansia muciniphila played a relevant role in infant microbiota [
16], in agreement with the high number of MAGs recovered from these clades.
It should be noted that some MAGs were identified at strain level, including
Blautia sp. SC05B48 (n = 38),
Ruminococcus sp. JE7A12 (n = 32),
Lachnoclostridium sp. YL32 (n = 25),
Longibaculum sp. KGMB06250 (n = 25),
Caproiciproducens sp. NJN-50 (n = 19),
Blautia sp. N6H1-15 (n = 16), Lachnospiraceae bacterium Choco86 (n = 16), Erysipelotrichaceae bacterium GAM147 (n = 15),
Streptococcus sp. HSISS3 (n = 11) and
Enterococcus sp. HSIEG1 (n = 8). Interestingly, metabolic pathways of Enterobacteriaceae and Lachnospiraceae families as well as
Blautia species had been also identified in infant metagenomes using assembly-free methods [
16].
Furthermore, up to 11 MAGs could be recovered from
Lacticaseibacillus rhamnosus, although few MAGs were obtained from other lactobacilli species. On the other hand,
Clostridium bolteae (n = 36) was the most frequent
Clostridium species, while a low number of MAGs could be obtained from other genera like
Bacillus,
Staphylococcus and
Streptococcus. Interestingly,
Clostridium genes were also identified in the assembly-free analysis of infant metagenomes [
16].
Concerning the sample origin of each MAG, most MAGs were recovered from formula-fed infants (n = 638), adults following fiber-rich dietary interventions (n = 467) and infant formula + GOS-fed infants (n = 281). Similarly, most bifidobacteria MAGs were obtained from formula-fed infants (n = 64), infant formula + GOS-fed infants (n = 52) and breastfed infants (n = 31), while only 11 bifidobacteria MAGs could be obtained from adult participants (
Supplementary Materials Table S2). Among these samples, most MAGs from
B. adolescentis were recovered from adults following fiber-rich dietary interventions (n = 8), while most MAGs from
B. longum and
B. pseudocatenulatum were recovered from infant formula-fed infants (n = 32 and n = 13, respectively). Similarly, most MAGs from
B. bifidum and
B. breve were recovered from breastfed (n = 11) and infant formula + GOS-fed infants (n = 16), respectively.
To deepen the metabolic potential of the MAGs species and strains obtained in this study and their variation with prebiotics consumption, glycosidase activities codified in MAGs were annotated, finding up to 80,028 functional domains belonging to 54 CAZy families of interest. These CAZy families were chosen based on their potential ability to metabolize the most common types of prebiotics (α- and β-GOS and FOS commonly added to infant formula), as well as HMOs. It must be considered that these oligosaccharide structures were present in the diet of most individuals considered in this study [
16]. Specific glycosidase activities aimed at hydrolyzing these substrates were selected to find characteristic metabolic profiles of the
Bifidobacterium genus depending on prebiotic consumption. Moreover, enzymatic profiles of prebiotic-exposed bifidobacterial MAGs were compared to those of bifidobacteria found in infants fed by non-supplemented formulas and adults exposed to high-fiber dietary interventions (and not specific prebiotic structures). Therefore, functional domains from CAZy families CBM32, CBM40, GH1-5, GH16, GH20, GH29, GH30-33, GH35-36, GH39, GH42, GH58-59, GH68, GH95, GH97, GH109-110, GH139, GH141, GH147 and GH151 involving α- and β-galactosidases, β-fructosyltransferases and β- fructofuranosidases, fucosidases, hexosaminidase and sialidases as well as other fiber-degrading activities were selected (
Supplementary Materials Table S3). Most metabolic activities reported during the assembly-free functional analysis of infant microbiota were limited to routes related to amino acid synthesis and degradation and vitamin synthesis pathways [
16]. In contrast, the present study deepens the characterization of carbohydrate-degrading enzymes present in these species, providing an extensive comparison of CAZy families found in
Bifidobacterium species and their ability to metabolize specific prebiotic structures.
3.2. Unsupervised Analysis to Study Glycosidase Distribution in Metagenome-Assembled Genomes (MAGs)
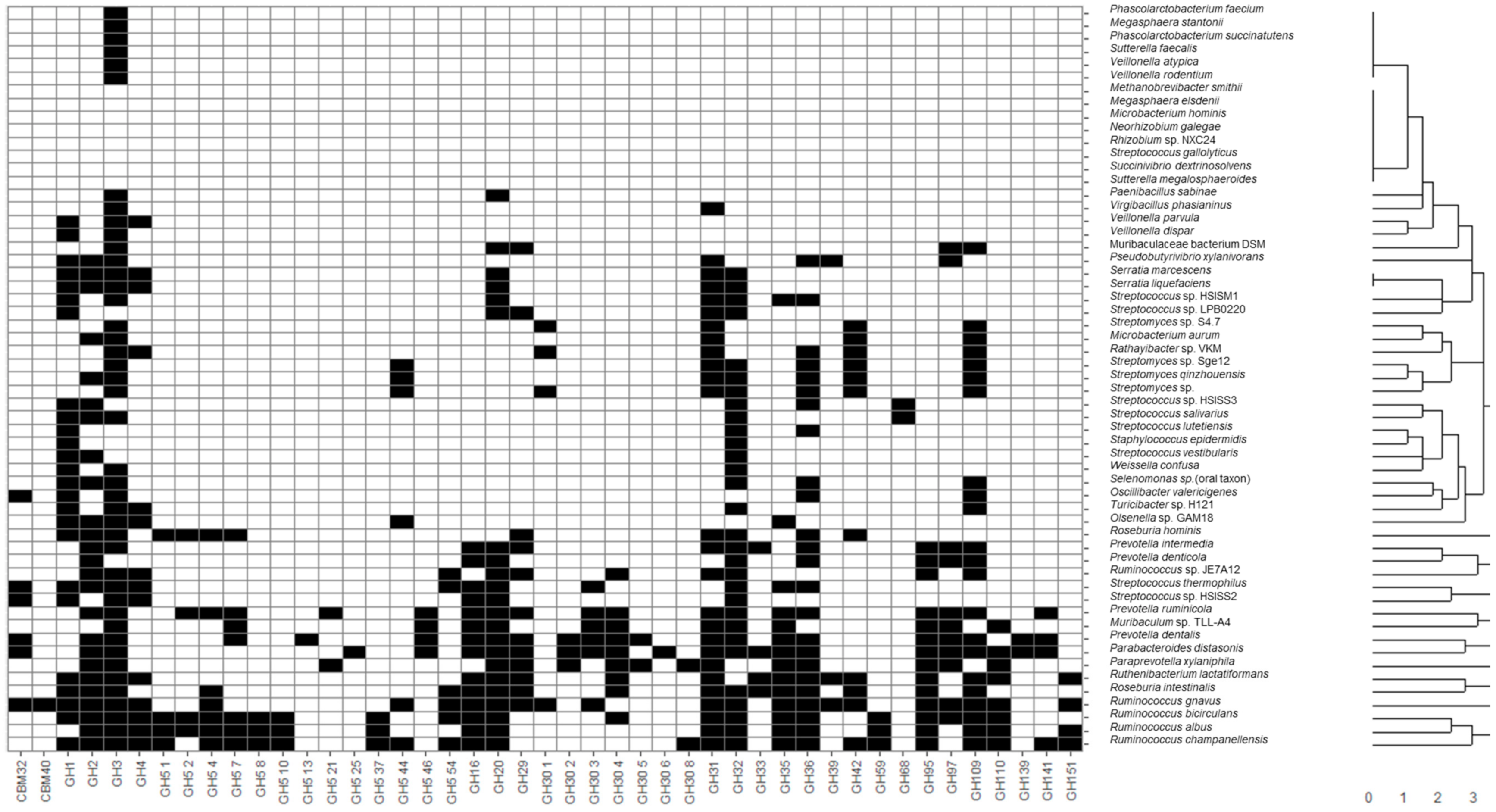
Recovered MAGs from all identified microbial species were clustered considering the presence of CAZy families of interest described in the previous section through hierarchical clustering considering a Euclidean distance metric (
Figure 1). Moreover, heatmaps illustrating the presence and absence of functional domains were generated. In this sense, applications of heatmaps to represent the presence or absence of microbial taxa and genes have been extensively reported in the literature. Some recent examples include the usage of heatmaps to elucidate the presence of coincident genes across microbial genomes [
34] and coincident genera in human microbiota samples [
35]. In the present study, most species belonging to the same genus were clustered together, highlighting common metabolic patterns. In general, the widest number of glycosidase activities codified in MAGs was observed for the following genera:
Bacteroides,
Blautia,
Caproiciproducens,
Clostridium,
Longibaculum,
Paraprevotella,
Prevotella,
Roseburia,
Ruminococcus,
Ruthenibacterium,
Muribaculum, as well as several species belonging to Lachnospiraceae and Erysipelotrichaceae families (
Figure 1). It should be noted that
Bacteroides, belonging to Bacteroidetes phyla, is one of the most relevant clades in the microbiota of adult individuals due to particular metabolic capabilities that allow
Bacteroides to use a wide range of complex carbohydrates [
36]. Other Bacteroidetes, such as
Paraprevotella,
Prevotella,
Muribaculum, may play a similar role based on their glycosidase profiles. In addition,
Roseburia comprises another relevant genus of the Firmicutes phyla known to metabolize dietary polysaccharides [
17]. Other Firmicutes that exhibited a wide range of prebiotic-degrading enzymes include
Blautia,
Caproiciproducens,
Clostridium,
Longibaculum,
Ruminococcus and
Ruthenibacterium. It has been reported that the healthy gut microbiome is composed predominantly of the phyla Firmicutes and Bacteroidetes [
37], and our cluster analysis highlights the prevalent role of the glycosidic metabolic activities of these two phyla, showing a wider variety of glycosidases able to hydrolyze some of the most common types of prebiotics.
Glycosidase activities from these bacteria corresponded mainly to the CBM32, CBM40, GH1-5, GH31-42, GH95-110 CAZy families comprising most enzyme classes included in this study, such as α- and β-galactosidases, fucosidases, fructosidases, hexosaminidases and sialidases. On the contrary, other species not characteristic of a healthy infant microbiota, according to Baumann-Dudenhoeffer et al. [
16] and Hill et al. [
38], lack several GH5 and GH30 subgroups comprising hexosaminidases and fucosidases, indicating a limited potential ability to metabolize HMOs.
A wide number of glycosidase activities were also found for Bifidobacterium, similar to those of the above-mentioned non-bifidobacteria species with high metabolic capacities, belonging to Firmicutes and Bacteroidetes. Moreover, some glycosidases were characteristic of bifidobacteria (i.e., not present in most MAGs species recovered): GH30 5 fucosidases were present in B. bifidum, B. breve and B. longum, as well as other non-bifidobacteria species like B. producta, C. sp. enoides, C. saccharolyticum, P. dentalis and P. xylaniphila, and L. bacterium strain GAM79. Similarly, GH59 β-galactosidases were characteristic of B. breve, B. longum and B. pseudocatenulatum, but were also present in C. bolteae, C. sp. enoides, C. saccharolyticum, F. prausnitzii, R. albus, R. bicirculans and R. champanellensis, and strains L. sp. YL32 and L. bacterium GAM79. As it can be seen, some characteristic glycosidases from bifidobacterial were also found in the Clostridium and Ruminococcus genera and novel strains from Lachnospiraceae, highlighting metabolic similarities between these species and a higher potential to degrade HMOs and mucins when compared to other microorganisms.
Other non- bifidobacteria species showed a limited number of CAZy families of interest, more limited than those observed for bifidobacteria. In this sense, only GH3 and GH31, comprising hexosaminidases and α-galactosidases, were found in
Mageeibacillus indolicus, while GH3 was the only relevant domain analyzed in MAGs from
Phascolarctobacterium and
Megasphaera genera. The presence of α-galactosidases in these species could be of interest to metabolize α-GOS commonly added to infant formula due to their potential prebiotic effect [
6]. Interestingly, GH30 3 family involving fucosidases was the only relevant domain determined in
Alistipes communis MAGs. It should be considered that fucosidases have been described in species from the genus
Alistipes [
39].
Main glycosidase activities for several species and strains presented in this work were previously reported in adult metagenomes by other authors involving xylan 1,4-beta-xylosidase, glucan endo-1,3-beta-D-glucosidase, glucan 1,6-alpha-glucosidase, licheninase, and cellulase [
17]. However, no attempts to elucidate glycosidase profiles of common prebiotic structures like GOS and FOS were made. Therefore, the results herein presented provide complementary information to those already reported in the bibliography and may provide a foundational basis to estimate which prebiotic structures could be more fermentable by a given species.
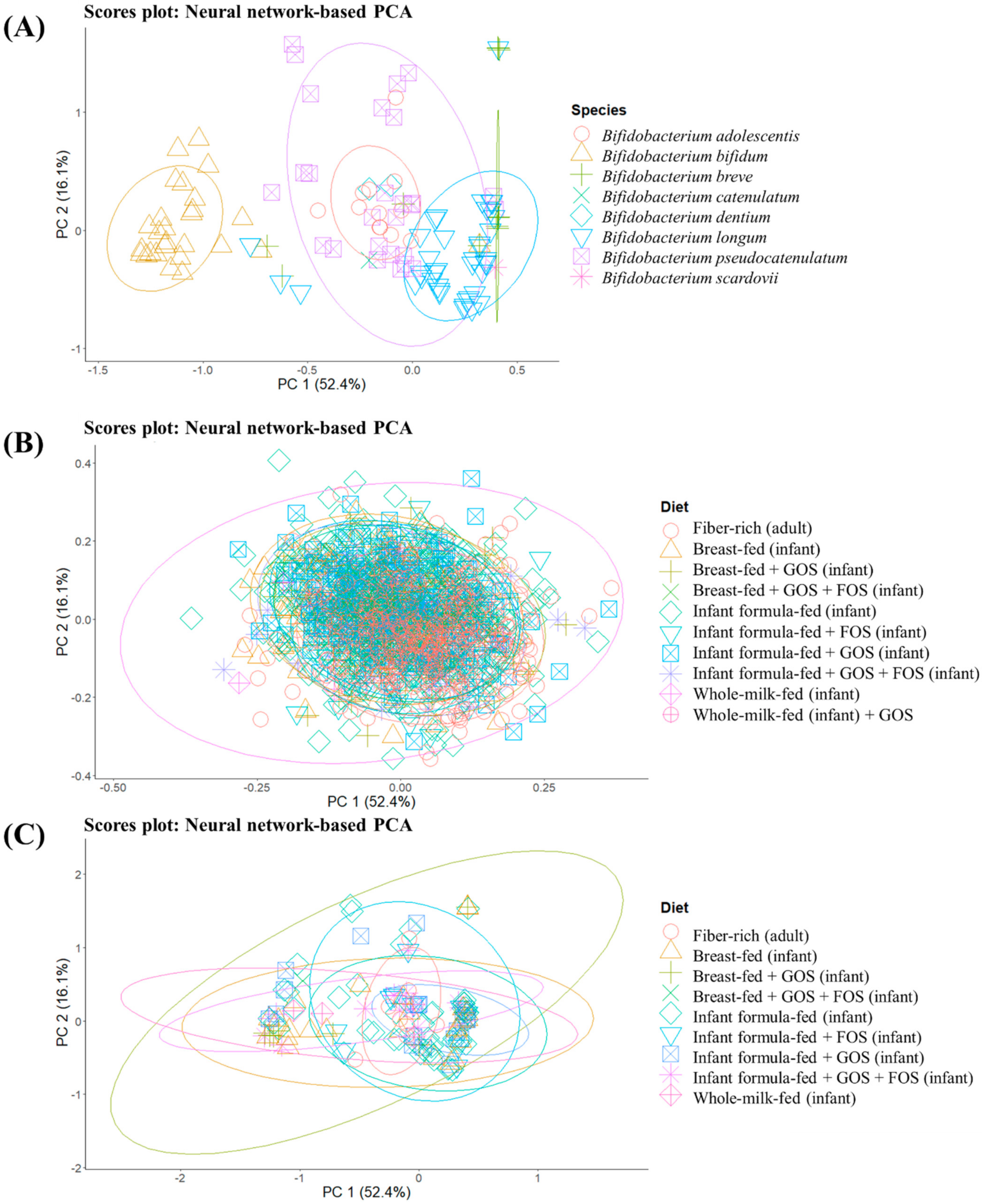
To get a general overview of glycosidase activities codified in different MAGs obtained from groups consuming different prebiotic structures, unsupervised sample distribution was evaluated by the ANN-based PCA model (
Figure 2). Specifically, differences between MAGs from different
Bifidobacterium species (
Figure 2A) as well as glycosidase profiles of MAGs from all identified species (and not just
Bifidobacterium,
Supplementary Materials Table S1) according to the host diet (
Figure 2B,C), are illustrated. The implementation of an ANN allows describing as much variance as possible, and the cumulative percentage of variance explained by the first five components was 89.2%, which could not be achieved by conventional PCA. Therefore, this kind of mathematical model could be especially suitable to find patterns in biological samples, which may yield sparse and heterogeneous data [
26,
27].
B. bifidum showed a characteristic glycosidic profile different from the rest of bifidobacteria (
B. adolescentis,
B. breve,
B. catenulatum,
B. dentium,
B. longum,
B. pseudocatenulatum,
B. scardovii) (
Figure 2A). This could be related to the potential ability of this species to degrade mucins [
40]. In addition, glycosidase patterns of
B. longum were distinguished from those of
B. adolescentis, considering the absence of overlap in normal ellipses and the percentages of variance explained.
When interpreting these differences, it should be considered that metabolic activities of
B. bifidum and
B. longum subsp.
infantis are tailored toward HMOs degradation, while other bifidobacteria do not encode the same HMOs-specific glycosidases and can degrade only limited HMOs [
41]. In contrast, no characteristic pattern could be elucidated when grouping MAGs from bifidobacteria and other species identified (
Figure 2B,C) according to the type of diet (breastfed, breastfed + GOS, breastfed + GOS + FOS, infant formula-fed, infant formula-fed + FOS, infant formula-fed + GOS, infant formula-fed + GOS + FOS and whole-milk-fed infants as well as fiber-rich diets in adults).
These results indicate the existence of different glycosidase profiles between Bifidobacterium species regardless of prebiotic exposure.
3.3. Supervised Classification to Establish Characteristic Glycosidase Profiles of Bifidobacterium Species
To deepen the study of characteristic glycosidases found in
Bifidobacterium MAGs, several supervised classification algorithms were compared, allowing establishing more robust patterns than the ones suggested by unsupervised projection. The number of MAGs recovered from
B. catenulatum,
B. dentium and
B. scardovii was not enough to train supervised algorithms (
Supplementary Materials Table S1), so these models were focused on
B. adolescentis,
B. bifidum,
B. breve,
B. longum,
B. pseudocatenulatum. ANN, RF and glmnet were trained on 70% of MAGs from each
Bifidobacterium species and tested on 30% new samples to ensure the reproducibility of the model.
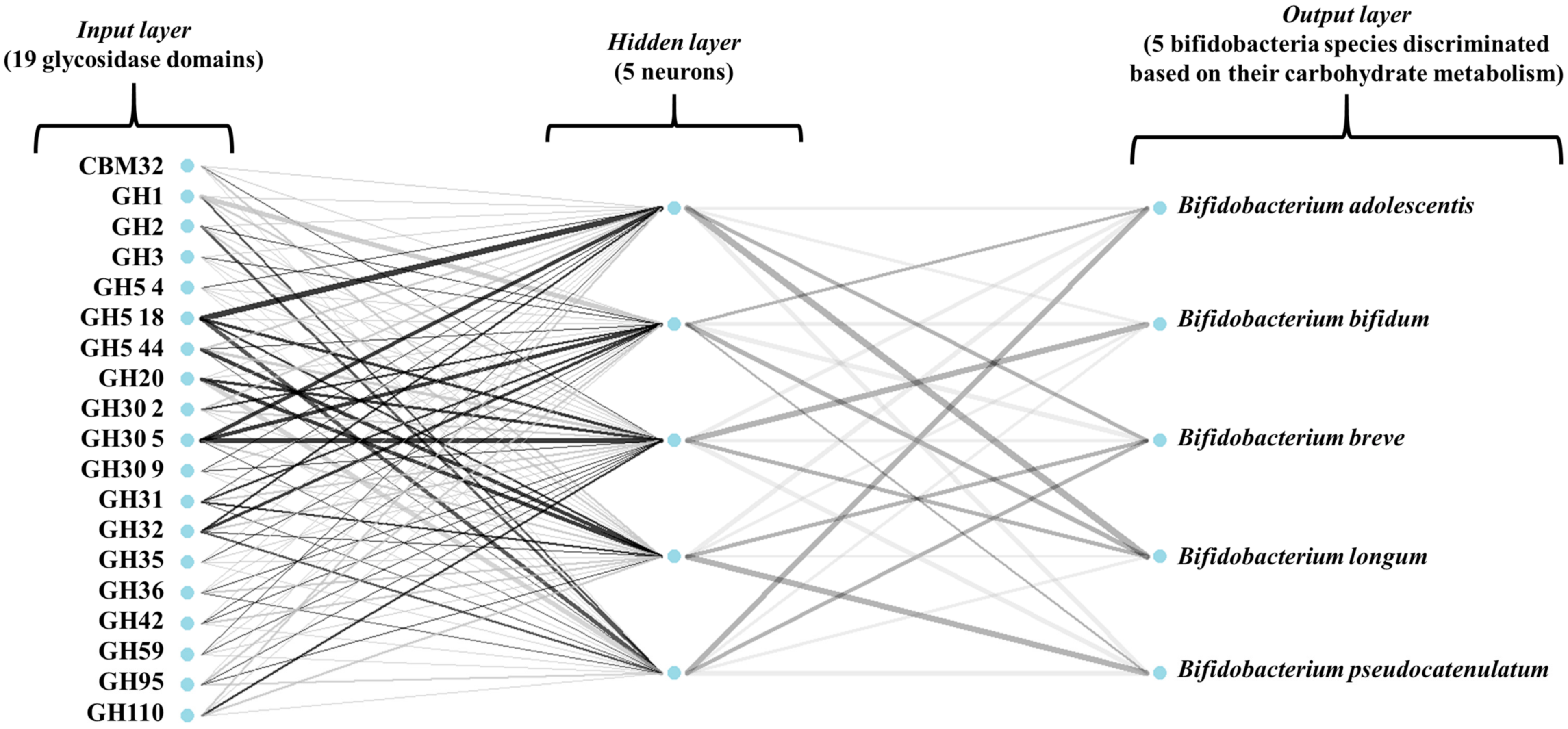
Figure 3 illustrates the architecture of the ANN model used in this study. In addition, all models were 10-fold cross-validated. To assess model performance, several estimators were calculated (
Supplementary Materials Table S4). The number of correctly classified MAGs during train and test phases was 97.8 and 91.7% for all models, while cross-validation accuracies for ANN, RF and glmnet were 96.4, 96.4 and 97.1%, respectively. Cross-validation kappa values, a more robust measure of accuracy that takes into account the possibility of correct classification by chance, for ANN, RF and glmnet, were 95.2, 96.2 and 96.2%, respectively. Similarly, kappa values obtained during the test phases were 89.2, 89.1 and 89.1% for ANN, RF and glmnet. A comparative account of the models was performed (
Supplementary Materials Figure S1), highlighting the absence of statistically significant differences (
p > 0.05) accuracies and kappa values calculated from the resampling distributions of the three algorithms. Additional estimators calculated include model sensitivity, specificity, precision, recall, F1, and balanced accuracy during the test phase on new samples.
Sensitivity, defined as the coefficient of the number of true-positive results by the total number of positives (including false-positives), recall, defined as the coefficient of true positives between relevant elements (i.e., MAGs from the same bifidobacteria species), and F1 coefficient, which combines precision and recall in one metric, showed lower values for
B. adolescentis (ranging from 0.63–0.83) than the rest of bifidobacteria in all models. This fact indicates a lower classification rate for
B. adolescentis than the rest of
Bifidobacterium species, revealing the absence of a unique glycosidase pattern for this species. As a consequence, characteristic glycosidases of
B. adolescentis are also characteristic of other bifidobacteria. However, specificity calculated by dividing the number of true-negative results by the total number of negatives (including false negatives) presented high values for all species, above 0.95 (
Supplementary Materials Table S4). Interestingly, precision, defined as the percentage of the model’s positive predictions that are accurate, showed the lowest values for
B. pseudocatenulatum (0.82 for the three models). As a consequence,
B. adolescentis and
B.
pseudocatenulatum exhibited the lowest balanced accuracies (0.81–0.86 and 0.93, respectively), defined as the sum of sensitivity and specificity divided by two.
These results could be attributed to minor differences existing in the glycosidase profiles of
B. adolescentis and
B. pseudocatenulatum recovered from infant or adult metagenomes and metabolic similarities between these two species, leading to misclassification of certain MAGs. Indeed, these two species belong to the same phylogenetic group [
42]. Machine learning trained MAG glycosidase data showed similar performance metrics than previous models trained on carbohydrate spectral data, highlighting the suitability of this mathematical approach to elucidate complex patterns within the field of probiotics and prebiotics [
27].
When interpreting these models, it should be considered that RF, glmnet and MLP are computed differently, although they yielded similar results, highlighting the characteristic activities of each Bifidobacterium species. These profiles were elucidated by complementary approaches, and the results from predictive algorithms reinforce each other. The unsupervised method could not properly discriminate between all Bifidobacterium species showing a lower performance (low percentages of variance explained by the first components). In contrast, supervised ANN, RF and MLP showed high classification accuracies, above 90% when tested on new samples. This fact may be attributed to the subtle differences existing between glycosidase domains found in MAGs that could not be properly explained by PCA-like methods. Therefore, advanced supervised pattern recognition methods are needed to elucidate the metabolic profiles of bifidobacteria.
To determine the most relevant glycosidase activities in the characteristic profile of
Bifidobacterium species, a variable importance analysis of each model was carried out (
Supplementary Materials Tables S5–S7). The most dominant glycosidase family from
B. adolescentis was GH2 (involving β-galactosidases). The absence of GH20 (comprising hexosaminidases) was also characteristic of this species, although
B. adolescentis shared the rest of its characteristic glycosidase domains with other
Bifidobacterium species, leading to a lower classification rate (
Supplementary Materials Table S4). Pokusaeva et al. [
43] suggested that
B. adolescentis is incapable of properly degrading HMOs, agreeing with our findings.
Relevant families in
B. bifidum profiles include GH5 44, GH32, and GH110 (hexosaminidases, fructosidases, fructosyl transferases and α-galactosidases). It should be noted that the GH110 CAZy family was not found in MAGs from other bifidobacteria (
Figure 1), indicating a different hydrolytic profile for
B. bifidum that may explain metabolic differences observed in PCA analysis (
Figure 2A). The role of hexosaminidases like β-N-acetylglucosaminidases on HMOs metabolism has been described by Sakanaka et al. [
2]. Specifically, the presence of hexosaminidases reported in
B. bifidum, B. breve and
B. longum subsp.
infantis genomes has been reported. Hexosaminidases from
B. bifidum, showing high importance coefficients in our machine learning models, could be highly active on HMOs like lacto-N-triose (LNTri), while those from other species like
B. breve could be active on lacto-N-tetraose (LNT) and lacto-N-neotetraose (LNnT). It has also been reported that
B. bifidum shows a high HMOs assimilation ability that may contribute to its characteristic glycosidase profile that favors its persistence in the breastfed infant gut [
2,
43]. In fact, previous studies indicate that
B. bifidum degrades some HMOs structures more rapidly than lactose [
43]. After host glycans and HMOs degradation, mono– or disaccharides released are consumed by other species metabolically dependent on these simple sugars [
4,
44].
Glycosidase profiles of
B. breve were characterized by GH1 and GH5 18 families (comprising β-galactosidases and hexosaminidases hydrolyzing mainly LNT and LNnT in contrast to those from
B. bifidum), and those from
B. longum showed high importance coefficients for β-galactosidases and fucosidases from GH1 and GH30 5 families. Finally, the presence of the glycosidase families, GH5 44 and GH20 comprising hexosaminidases and the absence of GH5 18 were characteristic traits from
B. pseudocatenulatum. These enzymes may contribute to the metabolism of lacto-N-biose I (LNB), according to previous authors [
2,
45].
We have demonstrated that it is possible to get highly accurate classifications of glycosidase activities from some of the most common Bifidobacterium species based on several mathematical approaches. Specifically, ANN, RF and glmnet exhibited high-performance metrics, indicating a high predictive power. As explained, characteristic glycosidase profiles of each bifidobacteria have been elucidated based on the importance coefficients from the three machine learning models. These models could be generalized and applied to new genomes from bifidobacteria and related microorganisms in future studies.
Concerning the main limitations of this method, it is possible that some glycosidase domains of interest may be lost during MAG assembly. However, the study of bifidobacteria MAGs allows comparing metabolic profiles of Bifidobacterium associated with specific groups of individuals (i.e., following dietary interventions of interest) and assessing the metabolic complementarity between bifidobacteria and non-bifidobacteria species found in the same participant.
3.4. Correlation Networks to Elucidate Glycosidase Activities Commonly Associated
To study the associations between different glycosidase activities (i.e., which glycosidase families are usually encoded together in the same MAG), correlation network models were computed (
Figure 4 and
Figure 5). The first correlation model (
Figure 4) was built using glycosidase activities from all species identified (bifidobacteria or not). Some of the strongest correlations observed include the positive associations between GH1 and GH4 and between GH36 and GH42 (involving α- and β-galactosidases). Furthermore, GH139 was positively associated with GH30 6 and GH147 families (involving β-galactosidases and fucosidases), and the GH151 family was correlated to CBM40 and GH97 (involving α-galactosidases, fucosidases and sialidases). In this sense, GH5 44 was associated with GH30 1, GH16, GH5 18 and GH30 2 families and GH59 was positively correlated to GH30 9 and GH5 22 families (comprising β-galactosidases, fucosidases and hexosaminidases). Similarly, several CAZy families involving hexosaminidases showed positive correlations: GH5 37 was associated with GH5 2, GH5 4 and GH5 1, while GH5 1 was associated with GH5 8, and GH5 22 was associated with GH5 9. It should be noted that all negative correlations were weaker than the positive ones, and no relevant associations were observed.
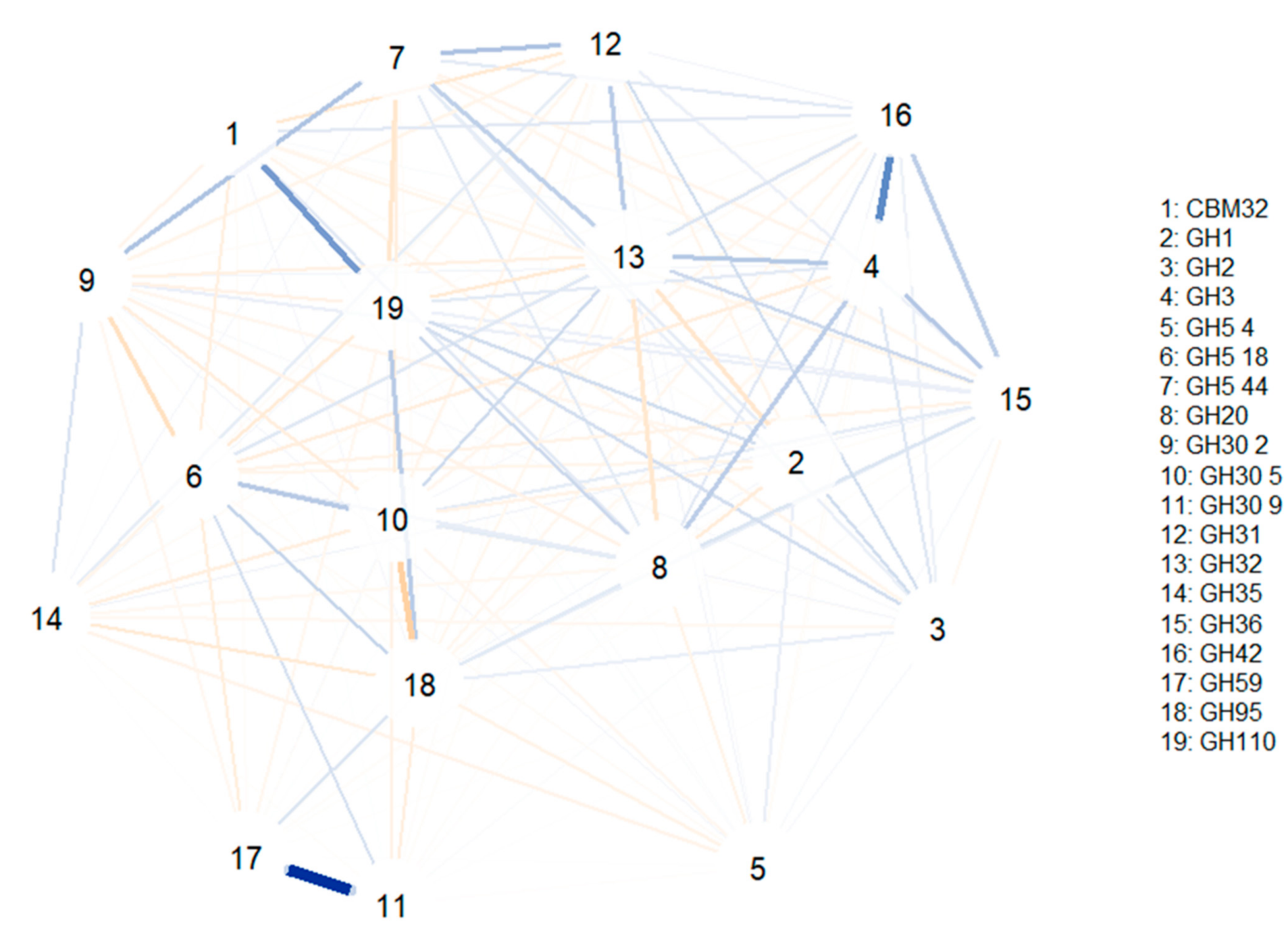
A second correlation network was computed to investigate glycosidase associations only in bifidobacteria MAGs (
Figure 5). GH30 9 and GH59 families involving fucosidase and β-galactosidase activities were positively associated, while a positive association was found between CBM32 and GH110 (comprising sialidases and α-galactosidases characteristic of
B. bifidum). In addition, GH3 was correlated to GH4; these two CAZy families comprising hexosaminidases and β-galactosidases. Similar to the previous study, no relevant negative correlations were observed.
As previously explained, fucosidases and β-galactosidases were strongly correlated not only in bifidobacteria but in all MAGs analyzed. It should be considered that these two enzyme families are involved in HMOs metabolism. Specifically, to degrade HMOs, both β-1,4-galactosidases and two types of fucosidases, 1,2-α-l-fucosidase and 1,3-1,4-α-l-fucosidase, are needed. Fucosidases were also correlated to sialidases, which may act on both α-2,3 and α-2,6 linkages found in sialylated HMOs [
2]. These results indicate that glycosidases that may potentially hydrolyze HMOs as well as glycans associated with mucin, sharing similar monomers, are strongly correlated and frequently found in the same MAG. Some HMOs-degrading enzymes were also strongly associated with the α-galactosidases characteristic of
B. bifidum, confirming the glycolytic profiles elucidated for this species in previous sections. It should be noted that α-galactosidases have been reported only in specific bifidobacterial species [
43] and may play a major in the metabolism of α-GOS like raffinose or stachyose, that are commonly added to infant formula [
6].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






