
Dynamics of Gut Microbiota Recovery after Antibiotic Exposure in Young and Old Mice (A Pilot Study)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Antibiotic Treatment
2.2. Gut Microbiota Analysis
3. Results
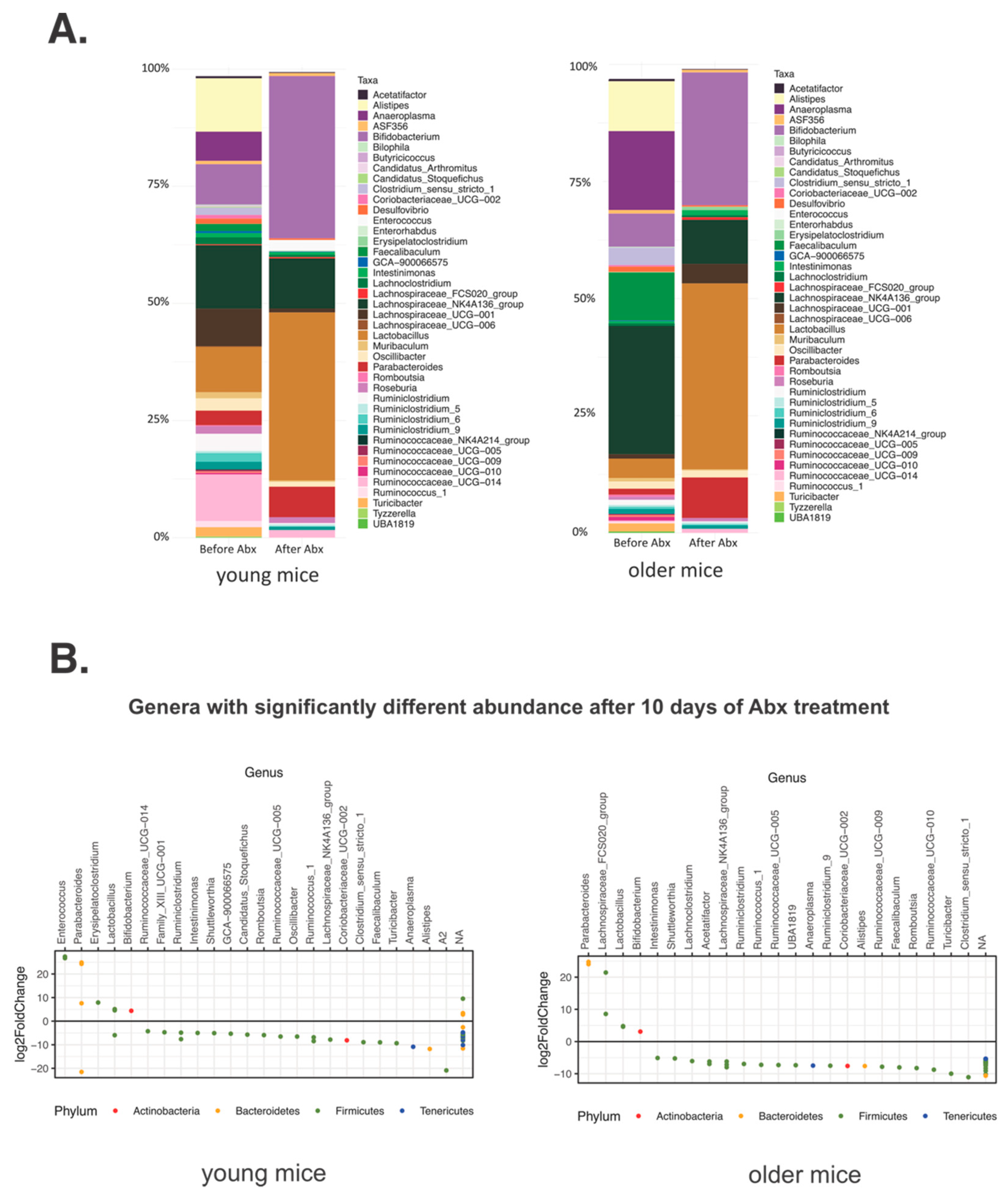
3.1. Short-Term Gut Microbiota Alterations after Broad-Spectrum Abx Treatment
3.2. Differential Long-Term Recovery from a Single Abx Treatment of Young and Older Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- De la Cuesta-Zuluaga, J.; Kelley, S.T.; Chen, Y.; Escobar, J.S.; Mueller, N.T.; Ley, R.E.; McDonald, D.; Huang, S.; Swafford, A.D.; Knight, R.; et al. Age- and Sex-Dependent Patterns of Gut Microbial Diversity in Human Adults. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Franceschi, C.; Rampelli, S.; Severgnini, M.; Ostan, R.; Turroni, S.; Consolandi, C.; Quercia, S.; Scurti, M.; Monti, D.; et al. Gut Microbiota and Extreme Longevity. Curr. Biol. 2016, 26, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, I.B.; Lynch, D.B.; O’Toole, P.W. Composition and temporal stability of the gut microbiota in older persons. ISME J. 2016, 10, 170–182. [Google Scholar] [CrossRef]
- Jackson, M.A.; Jeffery, I.B.; Beaumont, M.; Bell, J.T.; Clark, A.G.; Ley, R.E.; O’Toole, P.W.; Spector, T.D.; Steves, C.J. Signatures of early frailty in the gut microbiota. Genome Med. 2016, 8, 8. [Google Scholar] [CrossRef]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef]
- Borbet, T.C.; Zhang, X.; Muller, A.; Blaser, M.J. The role of the changing human microbiome in the asthma pandemic. J. Allergy Clin. Immunol. 2019, 144, 1457–1466. [Google Scholar] [CrossRef]
- Livanos, A.E.; Greiner, T.U.; Vangay, P.; Pathmasiri, W.; Stewart, D.; McRitchie, S.; Li, H.; Chung, J.; Sohn, J.; Kim, S.; et al. Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat. Microbiol. 2016, 1, 16140. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Chung, J.; Battaglia, T.; Henderson, N.; Jay, M.; Li, H.; Lieber, A.D.; Wu, F.; Perez-Perez, G.I.; Chen, Y.; et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 2016, 8, 343ra382. [Google Scholar] [CrossRef] [PubMed]
- Nobel, Y.R.; Cox, L.M.; Kirigin, F.F.; Bokulich, N.A.; Yamanishi, S.; Teitler, I.; Chung, J.; Sohn, J.; Barber, C.M.; Goldfarb, D.S.; et al. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat. Commun. 2015, 6, 7486. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, M.C.; Stiemsma, L.T.; Dimitriu, P.A.; Thorson, L.; Russell, S.; Yurist-Doutsch, S.; Kuzeljevic, B.; Gold, M.J.; Britton, H.M.; Lefebvre, D.L.; et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 2015, 7, 307ra152. [Google Scholar] [CrossRef] [PubMed]
- Korpela, K.; Salonen, A.; Saxen, H.; Nikkonen, A.; Peltola, V.; Jaakkola, T.; de Vos, W.; Kolho, K.-L. Antibiotics in early life associate with specific gut microbiota signatures in a prospective longitudinal infant cohort. Pediatr. Res. 2020, 88, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Ruohtula, T.; de Goffau, M.C.; Nieminen, J.K.; Honkanen, J.; Siljander, H.; Hamalainen, A.-M.; Peet, A.; Tillmann, V.; Ilonen, J.; Niemelä, O.; et al. Maturation of Gut Microbiota and Circulating Regulatory T Cells and Development of IgE Sensitization in Early Life. Front. Immunol. 2019, 10, 2494. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The microbiome in early life: Implications for health outcomes. Nat. Med. 2016, 22, 713–722. [Google Scholar] [CrossRef]
- Yassour, M.; Vatanen, T.; Siljander, H.; Hamalainen, A.M.; Harkonen, T.; Ryhanen, S.J.; Franzosa, E.A.; Vlamakis, H.; Huttenhower, C.; Gevers, D.; et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 2016, 8, 343ra381. [Google Scholar] [CrossRef]
- Dogra, S.; Sakwinska, O.; Soh, S.E.; Ngom-Bru, C.; Bruck, W.M.; Berger, B.; Brüssow, H.; Karnani, N.; Lee, Y.S.; Yap, F.; et al. Rate of establishing the gut microbiota in infancy has consequences for future health. Gut Microbes 2015, 6, 321–325. [Google Scholar] [CrossRef]
- Aleman, F.D.D.; Valenzano, D.R. Microbiome evolution during host aging. PLoS Pathog. 2019, 15, e1007727. [Google Scholar] [CrossRef]
- Li, H.; Qi, Y.; Jasper, H. Preventing Age-Related Decline of Gut Compartmentalization Limits Microbiota Dysbiosis and Extends Lifespan. Cell Host Microbe 2016, 19, 240–253. [Google Scholar] [CrossRef]
- Galley, J.D.; Bailey, M.T. Impact of stressor exposure on the interplay between commensal microbiota and host inflammation. Gut Microbes 2014, 5, 390–396. [Google Scholar] [CrossRef]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; de Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.G.; Fitzgerald, G.F.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4586–4591. [Google Scholar] [CrossRef]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkïla, J.; Monti, D.; Satokari, R.; Franceschi, C.; et al. Through ageing, and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Harrison, C.A.; Laubitz, D.; Midura-Kiela, M.T.; Jamwal, D.R.; Besselsen, D.G.; Ghishan, F.K.; Kiela, P.R. Sexual Dimorphism in the Response to Broad-spectrum Antibiotics During T Cell-mediated Colitis. J. Crohns Colitis 2019, 13, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Okansen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package. R package version 2.5-4. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 10 February 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef]
- Morgun, A.; Dzutsev, A.; Dong, X.; Greer, R.L.; Sexton, D.J.; Ravel, J.; Schuster, M.; Hsiao, W.; Matzinger, P.; Shulzhenko, N. Uncovering effects of antibiotics on the host and microbiota using transkingdom gene networks. Gut 2015, 64, 1732–1743. [Google Scholar] [CrossRef]
- Wei, S.; Mortensen, M.S.; Stokholm, J.; Brejnrod, A.D.; Thorsen, J.; Rasmussen, M.A.; Trivedi, U.; Bisgaard, H.; Sørensen, S.J. Short- and long-term impacts of azithromycin treatment on the gut microbiota in children: A double-blind, randomized, placebo-controlled trial. EBioMedicine 2018, 38, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Doan, T.; Arzika, A.M.; Ray, K.J.; Cotter, S.Y.; Kim, J.; Maliki, R.; Zhong, L.; Zhou, Z.; Porco, T.C.; Vanderschelden, B.; et al. Gut Microbial Diversity in Antibiotic-Naive Children After Systemic Antibiotic Exposure: A Randomized Controlled Trial. Clin. Infect. Dis. 2017, 64, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Korpela, K.; Salonen, A.; Virta, L.J.; Kekkonen, R.A.; Forslund, K.; Bork, P.; de Vos, W.M. Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat. Commun. 2016, 7, 10410. [Google Scholar] [CrossRef] [PubMed]
- Hicks, L.A.; Taylor, T.H., Jr.; Hunkler, R.J.U.S. outpatient antibiotic prescribing, 2010. N. Engl. J. Med. 2013, 368, 1461–1462. [Google Scholar] [CrossRef]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef]
- Fuchs, B.B.; Tharmalingam, N.; Mylonakis, E. Vulnerability of long-term care facility residents to Clostridium difficile infection due to microbiome disruptions. Future Microbiol. 2018, 13, 1537–1547. [Google Scholar] [CrossRef]
- Roubaud-Baudron, C.; Ruiz, V.E.; Swan, A.M., Jr.; Vallance, B.A.; Ozkul, C.; Pei, Z.; Li, J.; Battaglia, T.W.; Perez-Perez, G.I.; Blaser, M.J. Long-Term Effects of Early-Life Antibiotic Exposure on Resistance to Subsequent Bacterial Infection. mBio 2019, 10. [Google Scholar] [CrossRef]
- Patterson, A.M.; Mulder, I.E.; Travis, A.J.; Lan, A.; Cerf-Bensussan, N.; Gaboriau-Routhiau, V.; Garden, K.; Logan, E.; Delday, M.I.; Coutts, A.G.P.; et al. Human Gut Symbiont Roseburia hominis Promotes and Regulates Innate Immunity. Front. Immunol. 2017, 8, 1166. [Google Scholar] [CrossRef]
- Hearps, A.C.; Martin, G.E.; Angelovich, T.A.; Cheng, W.J.; Maisa, A.; Landay, A.L.; Jaworowski, A.; Crowe, S.M. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 2012, 11, 867–875. [Google Scholar] [CrossRef]
- Sovran, B.; Hugenholtz, F.; Elderman, M.; van Beek, A.A.; Graversen, K.; Huijskes, M.; Boekschoten, M.V.; Savelkoul, H.F.J.; de Vos, P.; Dekker, J.; et al. Age-associated Impairment of the Mucus Barrier Function is Associated with Profound Changes in Microbiota and Immunity. Sci. Rep. 2019, 9, 1437. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laubitz, D.; Typpo, K.; Midura-Kiela, M.; Brown, C.; Barberán, A.; Ghishan, F.K.; Kiela, P.R. Dynamics of Gut Microbiota Recovery after Antibiotic Exposure in Young and Old Mice (A Pilot Study). Microorganisms 2021, 9, 647. https://doi.org/10.3390/microorganisms9030647
Laubitz D, Typpo K, Midura-Kiela M, Brown C, Barberán A, Ghishan FK, Kiela PR. Dynamics of Gut Microbiota Recovery after Antibiotic Exposure in Young and Old Mice (A Pilot Study). Microorganisms. 2021; 9(3):647. https://doi.org/10.3390/microorganisms9030647
Chicago/Turabian StyleLaubitz, Daniel, Katri Typpo, Monica Midura-Kiela, Clairessa Brown, Albert Barberán, Fayez K. Ghishan, and Pawel R. Kiela. 2021. "Dynamics of Gut Microbiota Recovery after Antibiotic Exposure in Young and Old Mice (A Pilot Study)" Microorganisms 9, no. 3: 647. https://doi.org/10.3390/microorganisms9030647
APA StyleLaubitz, D., Typpo, K., Midura-Kiela, M., Brown, C., Barberán, A., Ghishan, F. K., & Kiela, P. R. (2021). Dynamics of Gut Microbiota Recovery after Antibiotic Exposure in Young and Old Mice (A Pilot Study). Microorganisms, 9(3), 647. https://doi.org/10.3390/microorganisms9030647