
Novel Therapies of Hepatitis B and D

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
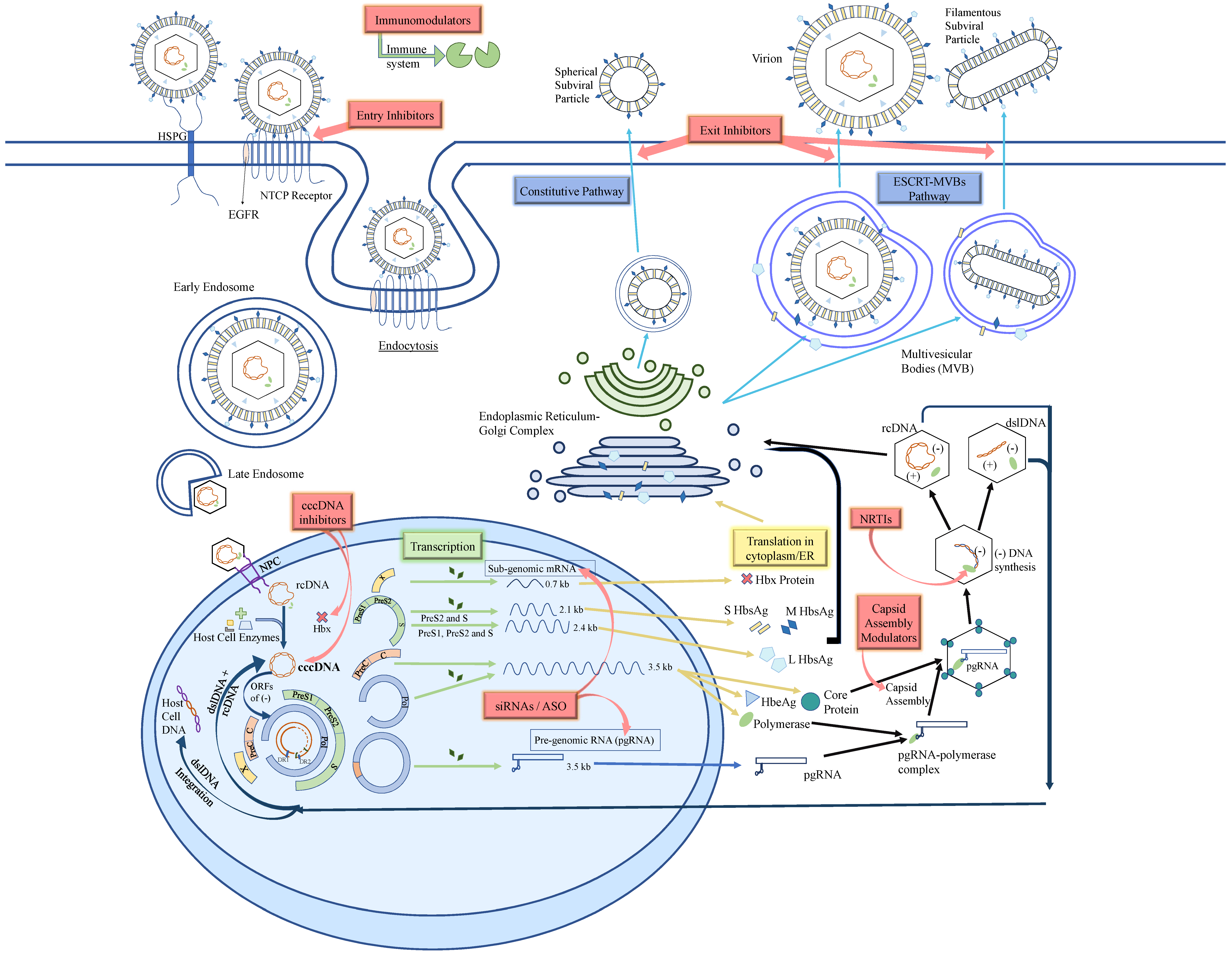
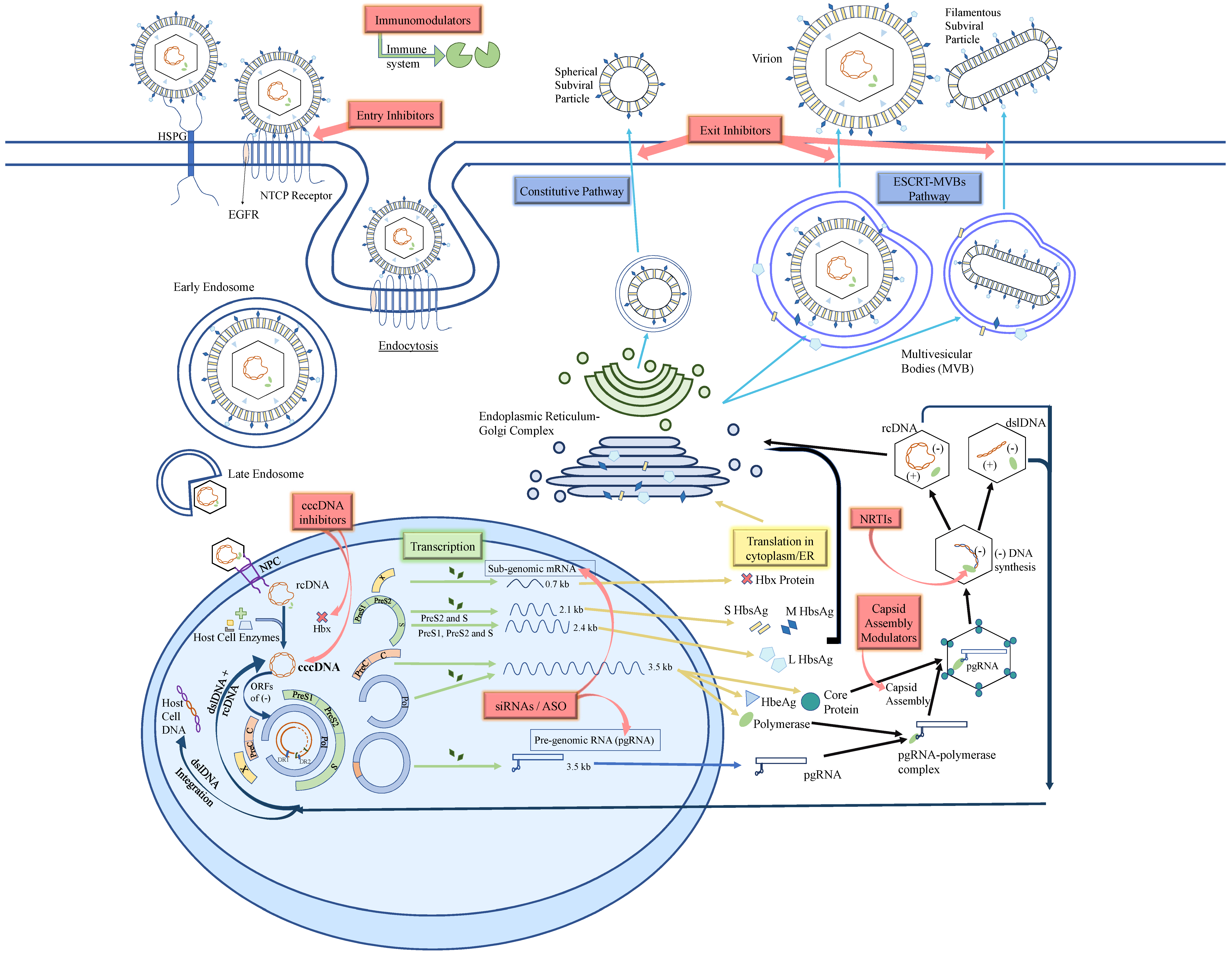
2. HBV Structure and Life Cycle
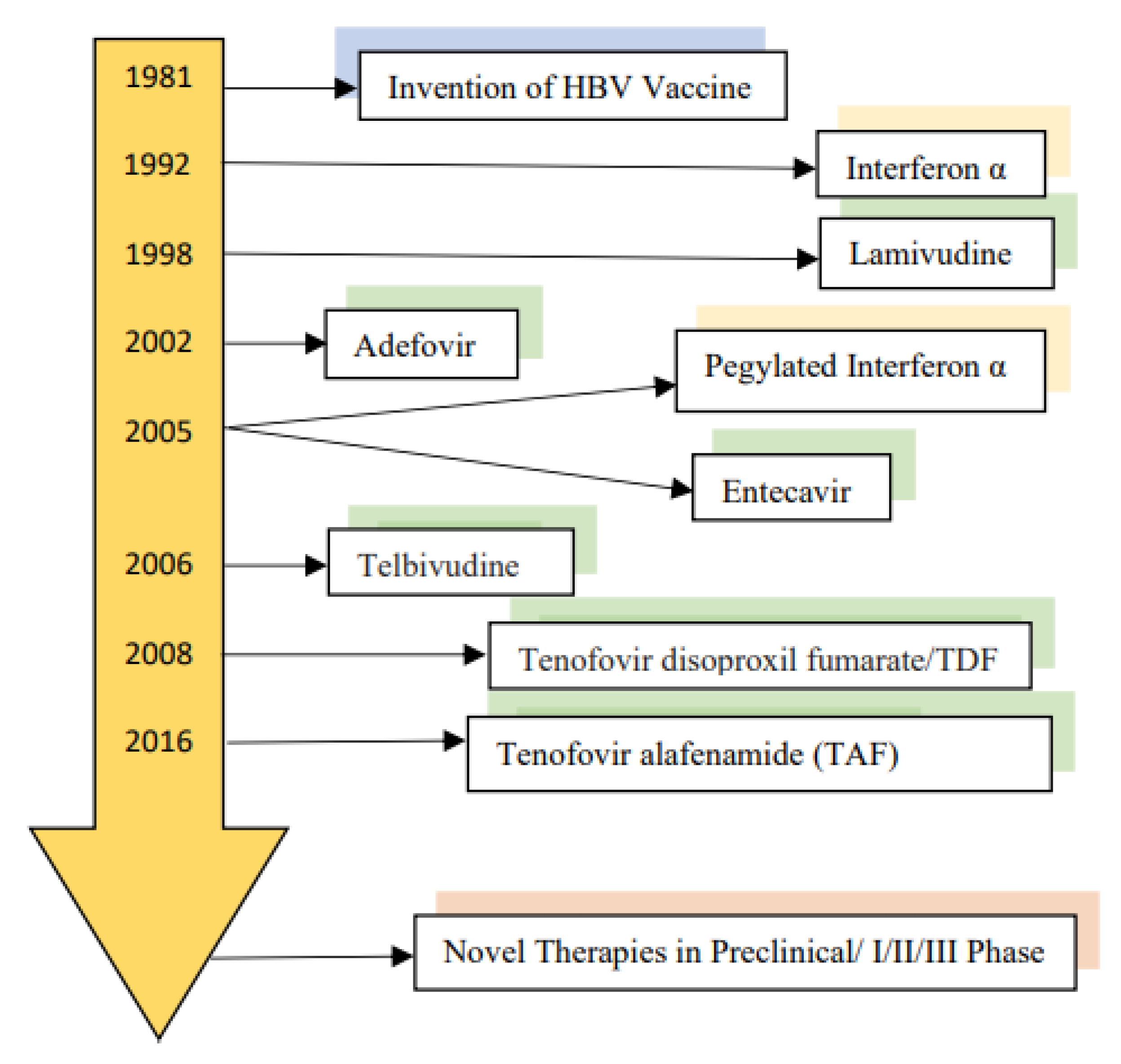
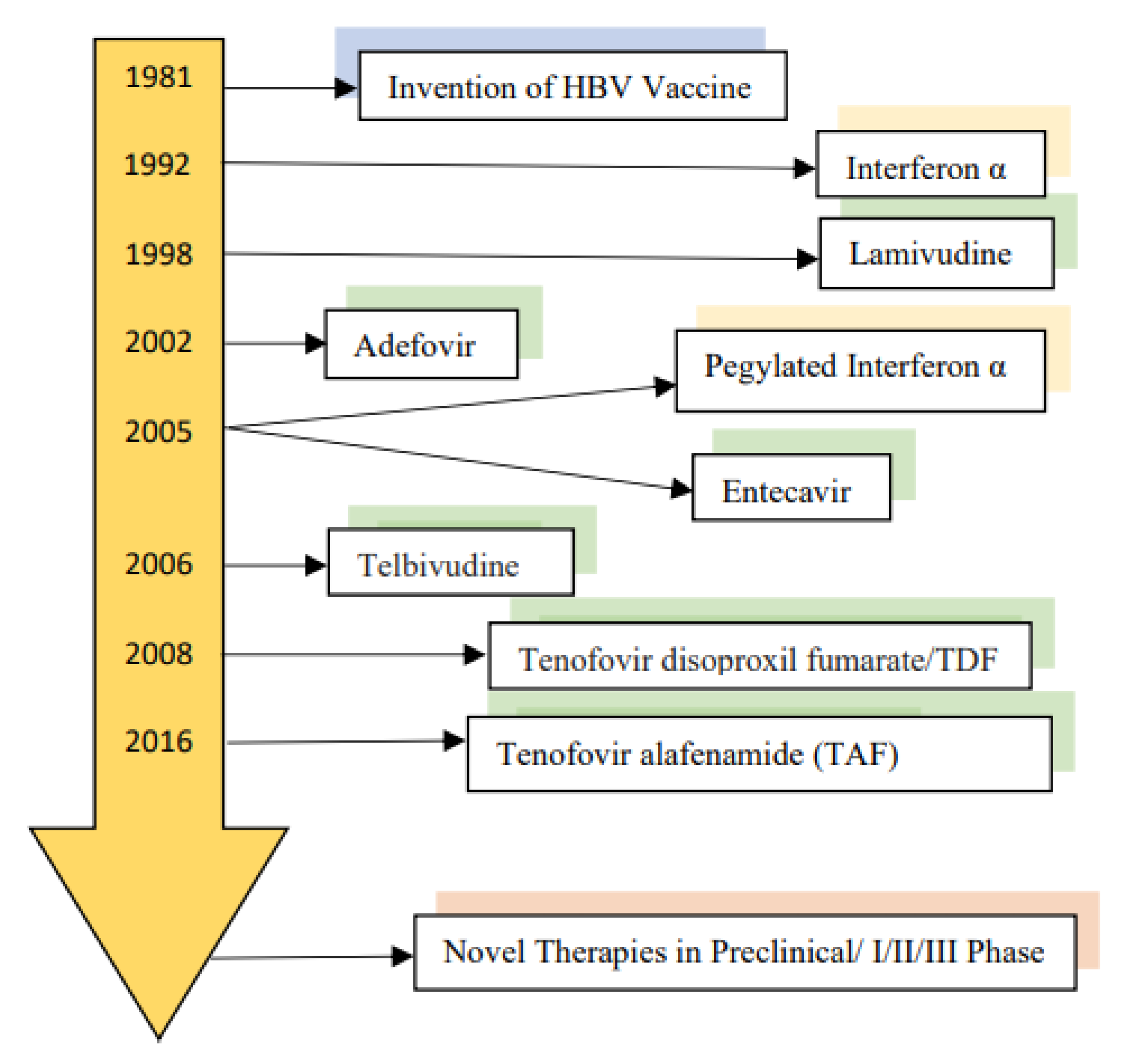
3. Current Therapy of Chronic Hepatitis B
4. Definition of HBV Cure
5. HBV Entry Inhibitor
6. cccDNA Inhibitors
6.1. CRISPR/Cas9
6.2. HBx Inhibitors
7. Small Interfering RNA (siRNA)
7.1. ARC 520
7.2. JN-3989 (ARO-HBV)
7.3. AB-729
7.4. VIR-2218
7.5. RG-6346
8. Anti-Sense Oligonucleotides (ASO)
8.1. GSK-3389404
8.2. GSK-3228836 (ISIS-505358)
8.3. RO-7062931 (LNA-SSO)
9. Core Protein Allosteric Modulators (CpAMs)
9.1. Types of CpAMs
9.1.1. Class 1 CpAM
R07049389
9.1.2. Class 2 CpAMs
ABI-HO731
NVR-3778
ABI-H2158
JNJ-6379
JNJ-0440
10. Nucleic Acid Polymers (NAPS)
11. Novel Immunomodulators
11.1. TLR 7 and TLR 8 Agonists
11.2. RIG-I Activator
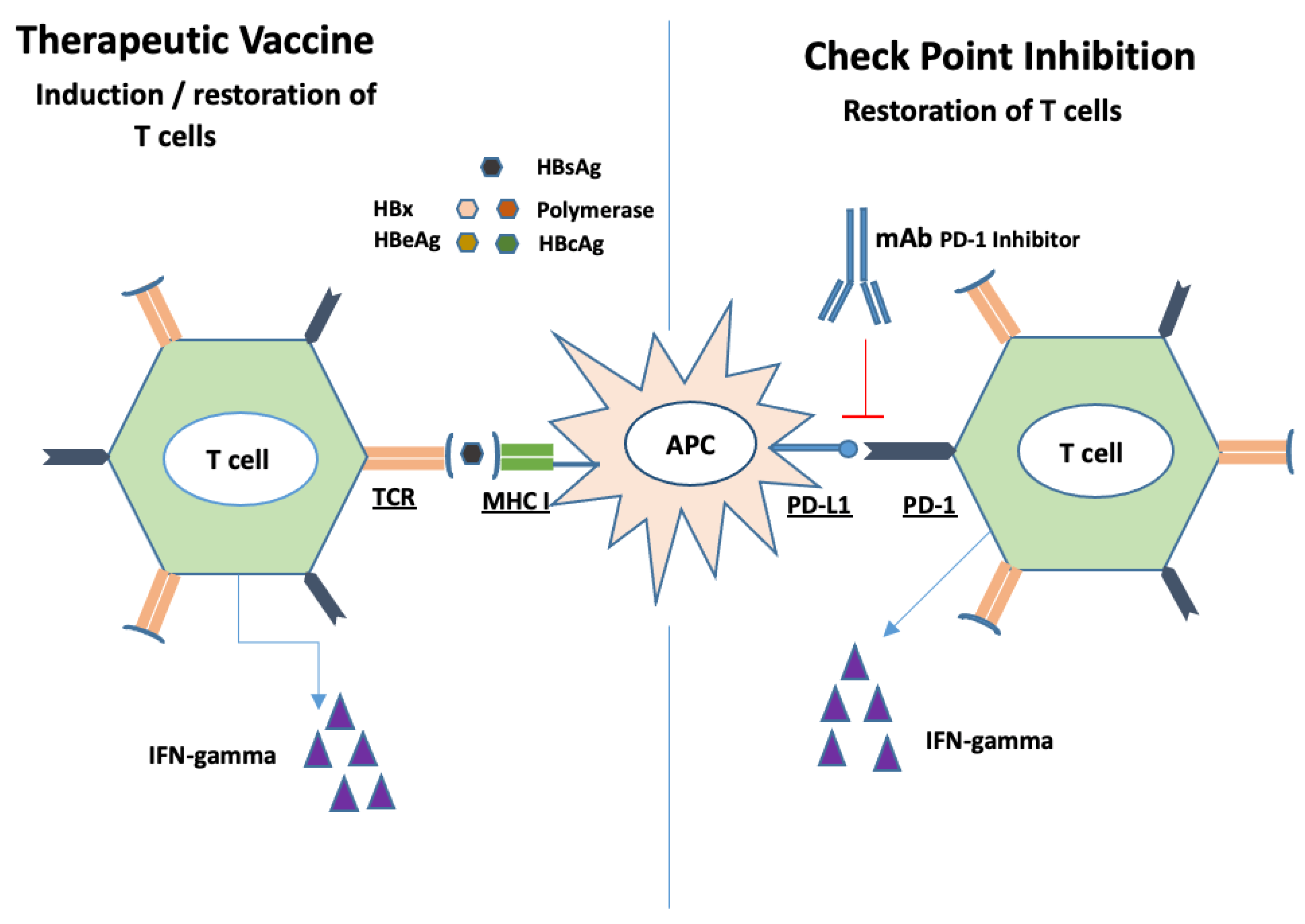
11.3. PD-1/PDL-1, CTLA4 Check Point Inhibitors
12. Engineered HBV Specific T-Cells
13. Therapeutic Vaccines
14. Novel HDV Therapies
14.1. PEG IFN-α
14.2. PEG IFN-LAMBDA (PEG IFN-λ)
14.3. Lonafarnib (LNF)
14.4. Entry Inhibitor
14.5. Nucleic Acid Polymers (NAPS)
15. Summary/Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization; Hepatitis, B. World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 1 October 2021).
- Xiong, Q.-F.; Xiong, T.; Huang, P.; Zhong, Y.-D.; Wang, H.-L.; Yang, Y.-F. Early predictors of acute hepatitis B progression to liver failure. PLoS ONE 2018, 13, e0201049. [Google Scholar] [CrossRef]
- McMahon, B.J. Epidemiology and natural history of hepatitis B. In Seminars in Liver Disease; Thieme Medical Publishers, Inc.: New York, NY, USA, 2005; Volume 25, pp. 3–8. [Google Scholar]
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. WJG 2014, 20, 5427–5434. [Google Scholar] [CrossRef]
- Lin, C.-L.; Kao, J.-H. Hepatitis B Virus Genotypes and Variants. Cold Spring Harb. Perspect. Med. 2015, 5, a021436. [Google Scholar] [CrossRef] [Green Version]
- Kramvis, A.; Kew, M.; François, G. Hepatitis B virus genotypes. Vaccine 2005, 23, 2409–2423. [Google Scholar] [CrossRef]
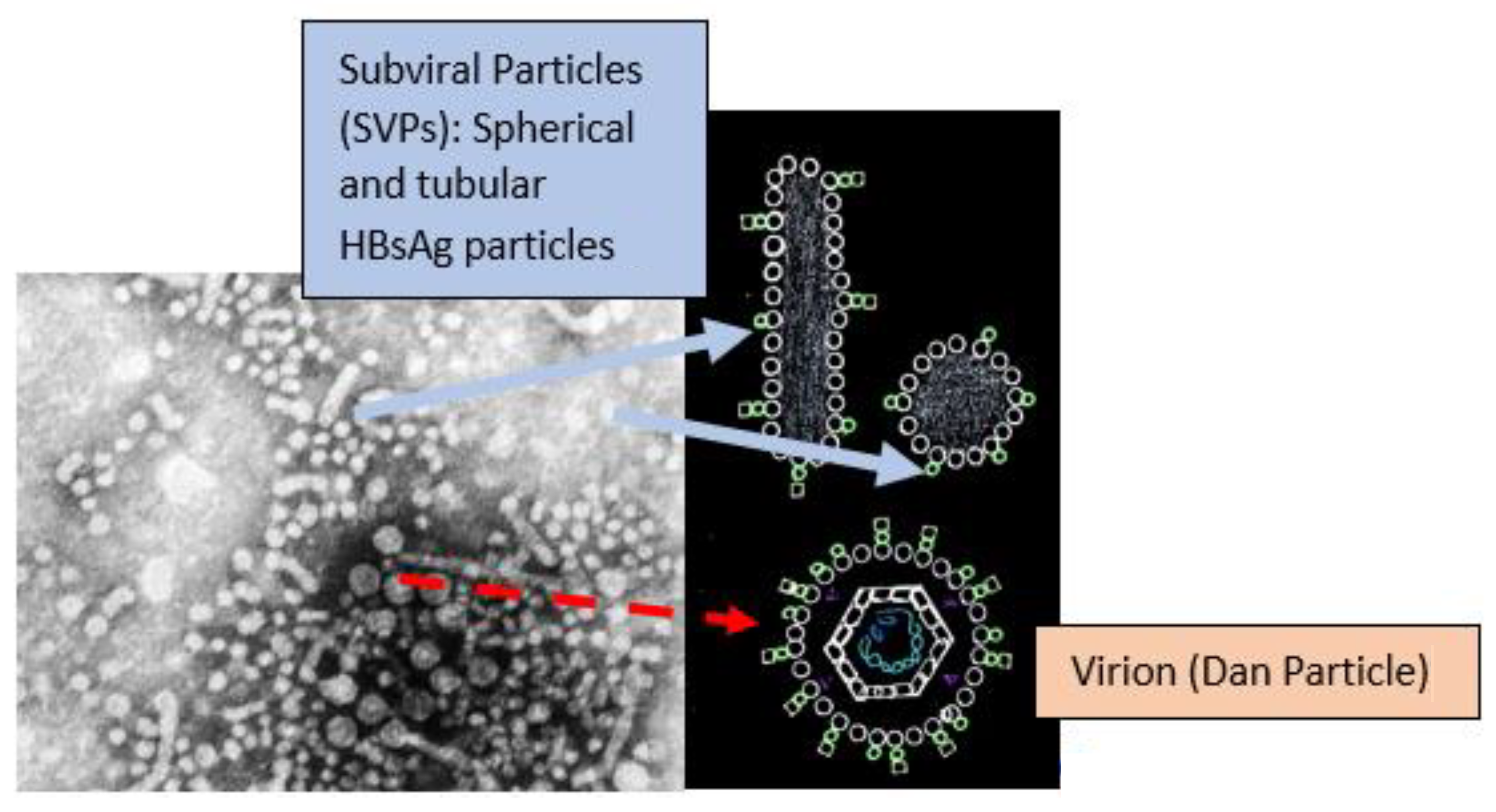
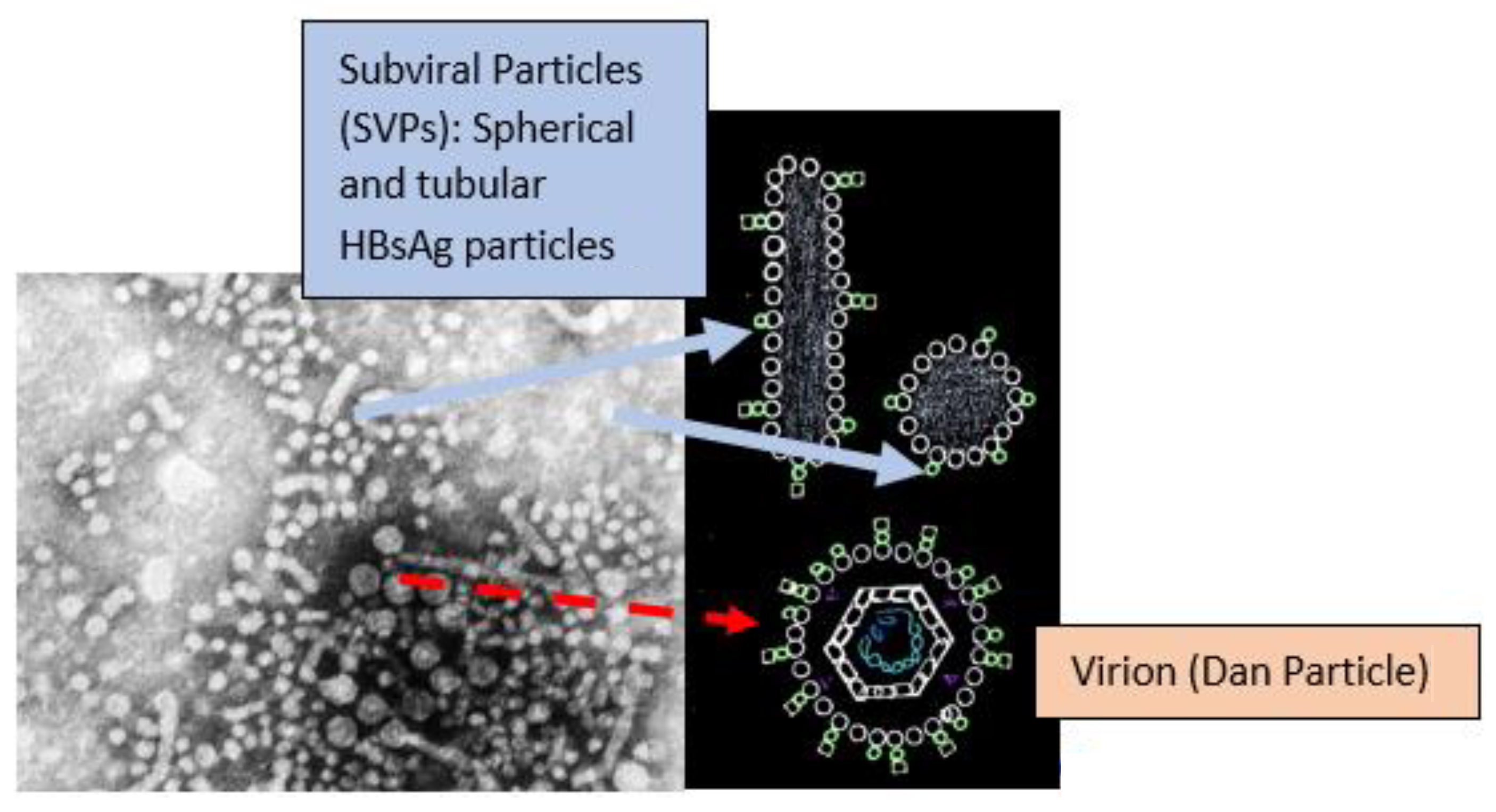
- Cao, J.; Zhang, J.; Lu, Y.; Luo, S.; Zhang, J.; Zhu, P. Cryo-EM structure of native spherical subviral particles isolated from HBV carriers. Virus Res. 2019, 259, 90–96. [Google Scholar] [CrossRef]
- Ho, J.K.-T.; Jeevan-Raj, B.; Netter, H.-J. Hepatitis B Virus (HBV) Subviral Particles as Protective Vaccines and Vaccine Platforms. Viruses 2020, 12, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, R.; Kottilil, S. Strategies to eliminate HBV infection. Future Virol. 2014, 9, 565–585. [Google Scholar] [CrossRef] [Green Version]
- Vaillant, A. HBsAg, Subviral Particles, and Their Clearance in Establishing a Functional Cure of Chronic Hepatitis B Virus Infection. ACS Infect. Dis. 2020, 7, 1351–1368. [Google Scholar] [CrossRef] [PubMed]
- Stannard, L.M.; Hodgkiss, M. Morphological Irregularities in Dane Particle Cores. J. Gen. Virol. 1979, 45, 509–514. [Google Scholar] [CrossRef]
- Boulon, R.; Blanchet, M.; Lemasson, M.; Vaillant, A.; Labonté, P. Characterization of the antiviral effects of REP 2139 on the HBV lifecycle in vitro. Antivir. Res. 2020, 183, 104853. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Allweiss, L.; Yang, D.; Kang, J.; Wang, J.; Qian, X.; Zhang, T.; Liu, H.; Wang, L.; Liu, S.; et al. Down-regulation of cell membrane localized NTCP expression in proliferating hepatocytes prevents hepatitis B virus infection. Emerg. Microbes Infect. 2019, 8, 879–894. [Google Scholar] [CrossRef]
- Herrscher, C.; Roingeard, P.; Blanchard, E. Hepatitis B Virus Entry into Cells. Cells 2020, 9, 1486. [Google Scholar] [CrossRef]
- Hu, J.; Cheng, J.; Tang, L.; Hu, Z.; Luo, Y.; Li, Y.; Zhou, T.; Chang, J.; Guo, J.-T. Virological Basis for the Cure of Chronic Hepatitis B. ACS Infect. Dis. 2018, 5, 659–674. [Google Scholar] [CrossRef]
- Gallucci, L.; Kann, M. Nuclear Import of Hepatitis B Virus Capsids and Genome. Viruses 2017, 9, 21. [Google Scholar] [CrossRef]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kang, H.S.; Kim, K.-H. Roles of hepatocyte nuclear factors in hepatitis B virus infection. World J. Gastroenterol. 2016, 22, 7017–7029. [Google Scholar] [CrossRef]
- Datta, S.; Chatterjee, S.; Veer, V.; Chakravarty, R. Molecular Biology of the Hepatitis B Virus for Clinicians. J. Clin. Exp. Hepatol. 2012, 2, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, B.; Brown, R. Strategies to Inhibit Hepatitis B Virus at the Transcript Level. Viruses 2021, 13, 1327. [Google Scholar] [CrossRef]
- Pollack, J.R.; Ganem, D. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J. Virol. 1993, 67, 3254–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.; Nassal, M. Efficient Hsp90-independent in Vitro Activation by Hsc70 and Hsp40 of Duck Hepatitis B Virus Reverse Transcriptase, an Assumed Hsp90 Client Protein. J. Biol. Chem. 2003, 278, 36128–36138. [Google Scholar] [CrossRef] [Green Version]
- Schädler, S.; Hildt, E. HBV Life Cycle: Entry and Morphogenesis. Viruses 2009, 1, 185–209. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.; Hildt, E. Intracellular Trafficking of HBV Particles. Cells 2020, 9, 2023. [Google Scholar] [CrossRef]
- Patient, R.; Hourioux, C.; Sizaret, P.-Y.; Trassard, S.; Sureau, C.; Roingeard, P. Hepatitis B Virus Subviral Envelope Particle Morphogenesis and Intracellular Trafficking. J. Virol. 2007, 81, 3842–3851. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Himmelsbach, K.; Ren, H.; Boller, K.; Hildt, E. Subviral Hepatitis B Virus Filaments, like Infectious Viral Particles, Are Released via Multivesicular Bodies. J. Virol. 2016, 90, 3330–3341. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.-H. Impact of hepatitis B vaccination on hepatitis B disease and nucleic acid testing in high-prevalence populations. J. Clin. Virol. 2006, 36, S45–S50. [Google Scholar] [CrossRef]
- Kruszon-Moran, D.; Paulose-Ram, R.; Martin, C.B.; Barker, L.K.; McQuillan, G. Prevalence and Trends in Hepatitis B Virus Infection in the United States, 2015–2018; NCHS Data Brief No. 361; National Center for Health Statistics: Hyattsville, MD, USA, 2020. [Google Scholar]
- Craxi, A.; Cooksley, W.G. Pegylated interferons for chronic hepatitis B. Antivir. Res. 2003, 60, 87–89. [Google Scholar] [CrossRef]
- Clark, D.N.; Hu, J. Hepatitis B virus reverse transcriptase—Target of current antiviral therapy and future drug development. Antivir. Res. 2015, 123, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Yim, H.J.; Kim, J.H.; Park, J.Y.; Yoon, E.L.; Park, H.; Kwon, J.H.; Sinn, D.H.; Lee, S.H.; Lee, J.-H.; Lee, H.W. Comparison of clinical practice guidelines for the management of chronic hepatitis B: When to start, when to change, and when to stop. Clin. Mol. Hepatol. 2020, 26, 411–429. [Google Scholar] [CrossRef]
- Lee, J.-H.; Cho, Y.; Lee, D.H.; Lee, M.; Yoo, J.-J.; Choi, W.-M.; Cho, Y.Y.; Bin Lee, Y.; Yu, S.J.; Yoon, J.-H.; et al. Prior Exposure to Lamivudine Increases Entecavir Resistance Risk in Chronic Hepatitis B Patients without Detectable Lamivudine Resistance. Antimicrob. Agents Chemother. 2014, 58, 1730–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, A.; Lin, X.; Chen, X. Functional cure for chronic hepatitis B: Accessibility, durability, and prognosis. Virol. J. 2021, 18, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Perrillo, R. Benefits and risks of interferon therapy for hepatitis B. Hepatology 2009, 49, S103–S111. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Ahn, S.H.; Ma, X.; Caruntu, F.A.; Tak, W.Y.; Elkashab, M.; Chuang, W.-L.; Lim, S.-G.; Tabak, F.; Mehta, R.; et al. Combination of Tenofovir Disoproxil Fumarate and Peginterferon α-2a Increases Loss of Hepatitis B Surface Antigen in Patients with Chronic Hepatitis B. Gastroenterology 2016, 150, 134–144.e10. [Google Scholar] [CrossRef] [Green Version]
- Viganò, M.; Mangia, G.; Lampertico, P. HB eAg-negative chronic hepatitis B: Why do I treat my patients with nucleos (t) ide analogues? Liver Int. 2014, 34, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Kranidioti, H.; Manolakopoulos, S.; Khakoo, S. Outcome after discontinuation of nucleot(s)ide analogues in chronic hepatitis B: Relapse rate and associated factors. Ann. Gastroenterol. Q. Publ. Hell. Soc. Gastroenterol. 2015, 28, 173–181. [Google Scholar]
- Cornberg, M.; Lok, A.S.; Terrault, N.A.; Zoulim, F.; Berg, T.; Brunetto, M.R.; Buchholz, S.; Buti, M.; Chan, H.L.; Chang, K.M.; et al. Guidance for design and endpoints of clinical trials in chronic hepatitis B-Report from the 2019 EASL-AASLD HBV Treatment Endpoints Conference. J. Hepatol. 2020, 72, 539–557. [Google Scholar] [CrossRef] [Green Version]
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.; Lau, D.T.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Primers 2018, 4, 1–20. [Google Scholar] [CrossRef]
- Schulze, A.; Schieck, A.; Ni, Y.; Mier, W.; Urban, S. Fine Mapping of Pre-S Sequence Requirements for Hepatitis B Virus Large Envelope Protein-Mediated Receptor Interaction. J. Virol. 2010, 84, 1989–2000. [Google Scholar] [CrossRef] [Green Version]
- Blank, A.; Markert, C.; Hohmann, N.; Carls, A.; Mikus, G.; Lehr, T.; Alexandrov, A.; Haag, M.; Schwab, M.; Urban, S.; et al. First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J. Hepatol. 2016, 65, 483–489. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M. Genome Editing Technologies as Cellular Defense Against Viral Pathogens. Front. Cell Dev. Biol. 2021, 9, 716344. [Google Scholar] [CrossRef]
- Chira, S.; Gulei, D.; Hajitou, A.; Zimta, A.A.; Cordelier, P.; Berindan-Neagoe, I. CRISPR/Cas9: Transcending the Reality of Genome Editing. Mol. Ther. Nucleic Acids 2017, 7, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.R.; Yang, H.C.; Kuo, Y.T.; Liu, C.J.; Yang, T.Y.; Sung, K.C.; Lin, Y.Y.; Wang, H.Y.; Wang, C.C.; Shen, Y.C.; et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol. Ther. Nucleic Acids 2014, 3, e186. [Google Scholar] [CrossRef]
- Stone, D.; Long, K.R.; Loprieno, M.A.; Feelixge, H.S.D.S.; Kenkel, E.J.; Liley, R.M.; Rapp, S.; Roychoudhury, P.; Nguyen, T.; Stensland, L.; et al. CRISPR-Cas9 gene editing of hepatitis B virus in chronically infected humanized mice. Mol. Ther. Methods Clin. Dev. 2021, 20, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Sekiba, K.; Otsuka, M.; Ohno, M.; Yamagami, M.; Kishikawa, T.; Suzuki, T.; Ishibashi, R.; Seimiya, T.; Tanaka, E.; Koike, K. Inhibition of HBV Transcription From cccDNA With Nitazoxanide by Targeting the HBx–DDB1 Interaction. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Sekiba, K.; Otsuka, M.; Ohno, M.; Yamagami, M.; Kishikawa, T.; Seimiya, T.; Suzuki, T.; Tanaka, E.; Ishibashi, R.; Funato, K.; et al. Pevonedistat, a Neuronal Precursor Cell-Expressed Developmentally Down-Regulated Protein 8–Activating Enzyme Inhibitor, Is a Potent Inhibitor of Hepatitis B Virus. Hepatology 2019, 69, 1903–1915. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 Mediates RNA Cleavage Targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Bock, C.T.; Wedemeyer, H.; Wüstefeld, T.; Locarnini, S.; Dienes, H.P.; Kubicka, S.; Manns, M.P.; Trautwein, C. Inhibition of hepatitis B virus replication in vivo by nucleoside analogues and siRNA. Gastroenterology 2003, 125, 9–18. [Google Scholar] [CrossRef]
- Konishi, M.; Wu, C.H.; Wu, G.Y. Inhibition of HBV replication by siRNA in a stable HBV-producing cell line. Hepatology 2003, 38, 842–850. [Google Scholar] [CrossRef]
- Dong, Y.; Siegwart, D.J.; Anderson, D.G. Strategies, design, and chemistry in siRNA delivery systems. Adv. Drug Deliv. Rev. 2019, 144, 133–147. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral Vectors Applied for RNAi-Based Antiviral Therapy. Viruses 2020, 12, 924. [Google Scholar] [CrossRef]
- Lok, A.S.; Zoulim, F.; Dusheiko, G.; Ghany, M.G. Hepatitis B cure: From discovery to regulatory approval. Hepatology 2017, 66, 1296–1313. [Google Scholar] [CrossRef]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Schluep, T.; Lickliter, J.; Hamilton, J.; Lewis, D.L.; Lai, C.-L.; Lau, J.Y.; Locarnini, S.A.; Gish, R.; Given, B.D. Safety, Tolerability, and Pharmacokinetics of ARC-520 Injection, an RNA Interference-Based Therapeutic for the Treatment of Chronic Hepatitis B Virus Infection, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2017, 6, 350–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooddell, C.I.; Yuen, M.F.; Chan, H.L.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees implicates integrated hepatitis B virus DNA as a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef] [Green Version]
- Yuen, M.-F.; Wong, D.K.-H.; Schluep, T.; Lai, C.-L.; Ferrari, C.; Locarnini, S.; Lo, R.C.-L.; Gish, R.G.; Hamilton, J.; Wooddell, C.I.; et al. Long-term serological, virological and histological responses to RNA inhibition by ARC-520 in Chinese chronic hepatitis B patients on entecavir treatment. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, F.; Limani, S.W.; Mnyandu, N.; Maepa, M.B.; Ely, A.; Arbuthnot, P. Advances with RNAi-based therapy for hepatitis B virus infection. Viruses 2020, 12, 851. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Locarnini, S.; Lim, T.H.; Strasser, S.; Sievert, W.; Cheng, W.; Thompson, A.; Given, B.; Schluep, T.; Hamilton, J.; et al. First results with RNA interference (RNAi) in chronic hepatitis B (CHB) using ARO-HBV. In American Association for the Study of Liver Diseases (AASLD): The Liver Meeting 2017 2018; John Wiley & Sons, Inc.: Hoboken, NJ, USA; Available online: https://aasldpubs.onlinelibrary.wiley.com/journal/15273350 (accessed on 1 October 2021).
- Gane, E.J.; Locarnini, S.; Lim, T.H.; Strasser, S.I.; Sievert, W.; Cheng, W.; Thompson, A.; Given, B.D.; Schluep, T.; Hamilton, J.; et al. Dose response with the RNA interference (RNAi) therapy JNJ-3989 combined with nucleos (t) ide analogue (NA) treatment in expanded cohorts of patients (PTS) with chronic hepatitis B (CHB). In Proceedings of the Poster abstract, AASLD The Liver Meeting, Boston, MA, USA, 9–13 November 2019. [Google Scholar]
- Gane, E.; Locarnini, S.; Lim, T.H.; Strasser, S.; Sievert, W.; Cheng, W.; Thompson, A.; Given, B.; Schluep, T.; Hamilton, J.; et al. Short-term treatment with RNA interference therapy, JNJ-3989, results in sustained hepatitis B surface antigen suppression in patients with chronic hepatitis B receiving nucleos (t) ide analogue treatment. In Proceedings of the Digital International Liver Congress 2020, Online, 27–29 August 2020. [Google Scholar]
- Yuen, M.F.; Berliba, E.; Kim, Y.J.; Holmes, J.A.; Lim, Y.S.; Strasser, S.I.; Schwabe, C.; Jucov, A.; Lee, A.C.; Thi, E.P.; et al. Safety and Pharmacodynamics of the GalNAc-siRNA AB-729 in subjects with Chronic Hepatitis B infection. In Hepatology; WILEY: Hoboken, NJ, USA, 2020; Volume 72, pp. 62A–63A. [Google Scholar]
- Gupta, S.V.; Fanget, M.C.; Bakardjiev, A.; Robbie, G.I.; Goel, V.; Mogalian, E. Pharmacokinetics of VIR-2218, an RNAi therapeutic for the treatment of HBV infection, in healthy volunteers. J. Hepatol. 2020, 73, S885. [Google Scholar] [CrossRef]
- Gane, E.; Lim, Y.S.; Tangkijvanich, P.; O’Beirne, J.; Lim, T.H.; Bakardjiev, A.; Ding, X.; Connolly, L.; Huang, S.; Kim, J.; et al. Preliminary safety and antiviral activity of VIR-2218, an X-targeting HBV RNAi therapeutic, in chronic hepatitis B patients. In Proceedings of the Digital International Liver Congress 2020, Online, 27–29 August 2020. [Google Scholar]
- Gane, E.; Lim, Y.S.; Cloutier, D.; Shen, L.; Cathcart, A.; Ding, X.; Pang, P.; Huang, S.; Yuen, M.F. Safety and antiviral activity of VIR-2218, an X-targeting RNAi therapeutic, in participants with chronic hepatitis B infection: Week 48 follow-up results. In Journal of Hepatology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 75, pp. S287–S288. [Google Scholar]
- Yuen, M.F.; Lim, Y.S.; Cloutier, D.; Shen, L.; Arizpe, A.; Pang, P.; Tay, C.; Thanawala, V.; Gupta, S.V.; Cathcart, A.; et al. Preliminary on-treatment data from a phase 2 study evaluating VIR-2218 in combination with pegylated interferon alfa-2a in participants with chronic hepatitis B infection. In Journal of Hepatology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 75, pp. S738–S739. [Google Scholar]
- Yuen, M.F.; Lim, T.H.; Kim, W.; Tongkijvonich, P.; Yoon, J.H.; Sievert, W.; Sukeepoisornjoroen, W.; Thompson, A.; Schwabe, C.; Brown, B.; et al. HBV RNAi inhibitor RG6346 in Phase 1b-2a trial was safe, well tolerated, and resulted in subatantial and durable reductions in serum HBsAg levels. In Proceedings of the AASLD 2020, Online, 13–16 November 2020. [Google Scholar]
- Han, K.; Cremer, J.; Elston, R.; Oliver, S.; Baptiste-Brown, S.; Chen, S.; Gardiner, D.; Davies, M.; Saunders, J.; Hamatake, R.; et al. A Randomized, Double-Blind, Placebo-Controlled, First-Time-in-Human Study to Assess the Safety, Tolerability, and Pharmacokinetics of Single and Multiple Ascending Doses of GSK3389404 in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2019, 8, 790–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, R.M.; Heo, J.; Kumada, H.; Suzuki, F.; Suzuki, Y.; Xie, Q.; Jia, J.D.; Karino, Y.; Hou, J.L.; Chayama, K.; et al. Results after 12 weeks treatment of multiple doses of GSK3389404 in chronic hepatitis B (CHB) subjects on stable nucleos (t) ide therapy in a phase 2a double-blind, placebo-controlled study. In Proceedings of the Poster Abstract, AASLD The Liver Meeting, Boston, MA, USA, 9–13 November 2019. [Google Scholar]
- Yuen, M.-F.; Heo, J.; Jang, J.W.; Yoon, J.-H.; Kweon, Y.O.; Park, S.-J.; Bennett, C.F.; Kwoh, T.J. Hepatitis B virus (HBV) surface antigen (HBsAg) inhibition with isis 505358 in chronic hepatitis B (CHB) patients on stable nucleos (t)ide analogue (NA) regimen and in NA-naive CHB patients: Phase 2a, randomized, double-blind, placebo-controlled study. In Proceedings of the Digital International Liver Congress 2020, Online, 27–29 August 2020. [Google Scholar] [CrossRef]
- Yuen, M.-F.; Gane, E.; Kim, D.J.; Chan, H.; Surujbally, B.; Pavlovic, V.; Triyatni, M.; Grippo, J.; Kim, H.J.; Leerapun, A.; et al. RO7062931 antisense oligonucleotide phase 1 study demonstrates target engagement in patients with chronic hepatitis B on established nucleos(t)ide therapy. In Proceedings of the Digital International Liver Congress 2020, Online, 27–29 August 2020. [Google Scholar] [CrossRef]
- Tsounis, E.P.; Tourkochristou, E.; Mouzaki, A.; Triantos, C. Toward a new era of hepatitis B virus therapeutics: The pursuit of a functional cure. World J. Gastroenterol. 2021, 27, 2727–2757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, J.; Ma, J.; Hu, Z.; Wu, S.; Hwang, N.; Kulp, J.; Du, Y.; Guo, J.T.; Chang, J. Discovery of novel hepatitis B virus nucleocapsid assembly inhibitors. ACS Infect. Dis. 2018, 5, 759–768. [Google Scholar] [CrossRef]
- Guo, F.; Zhao, Q.; Sheraz, M.; Cheng, J.; Qi, Y.; Su, Q.; Cuconati, A.; Wei, L.; Du, Y.; Li, W.; et al. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog. 2017, 13, e1006658. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wu, D.; Hu, H.; Zeng, J.; Yu, X.; Xu, Z.; Zhou, Z.; Zhou, X.; Yang, G.; Young, J.A.; et al. Direct Inhibition of Hepatitis B e Antigen by Core Protein Allosteric Modulator. Hepatology 2019, 70, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, R.W.-H.; Mak, L.-Y.; Seto, W.-K.; Yuen, M.-F. Role of Core/Capsid Inhibitors in Functional Cure Strategies for Chronic Hepatitis B. Curr. Hepatol. Rep. 2020, 19, 293–301. [Google Scholar] [CrossRef]
- Yuen, M.-F.; Zhou, X.; Gane, E.; Schwabe, C.; Tanwandee, T.; Feng, S.; Jin, Y.; Triyatni, M.; Lemenuel-Diot, A.; Cosson, V.; et al. Safety, pharmacokinetics, and antiviral activity of RO7049389, a core protein allosteric modulator, in patients with chronic hepatitis B virus infection: A multicentre, randomised, placebo-controlled, phase 1 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 723–732. [Google Scholar] [CrossRef]
- Yuen, M.-F.; Agarwal, K.; Gane, E.J.; Schwabe, C.; Ahn, S.H.; Kim, D.J.; Lim, Y.-S.; Cheng, W.; Sievert, W.; Visvanathan, K.; et al. Safety, pharmacokinetics, and antiviral effects of ABI-H0731, a hepatitis B virus core inhibitor: A randomised, placebo-controlled phase 1 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 152–166. [Google Scholar] [CrossRef]
- Sulkowski, M.; Agarwal, K.; Fung, S.; Yuen, M.F.; Ma, X.; Lalezari, J.; Nguyen, T.T.; Bae, H.; Schiff, E.R.; Hassanein, T.; et al. Continued therapy with ABI-H0731+ NrtI results in sequential reduction/loss of HBV DNA, HBV RNA, HBeAg, HBcrAg and HBsAg in HBeAg-positive patients. Hepatology 2019, 70, 1486A–1487A. [Google Scholar]
- Gane, E.; Sulkowski, M.; Ma, X.; Nguyen, T.; Hann, H.; Hassanein, T.; Elkhashab, M.; Nahass, R.; Chan, S.; Bennett, M. Viral response and safety following discontinuation of treatment with the core inhibitor vebicorvir and a nucleos (t) ide reverse transcriptase inhibitor in patients with HBeAg positive or negative chronic hepatitis B virus infection. J. Hepatol. 2021, 75, S736. [Google Scholar]
- Yuen, M.F.; Gane, E.J.; Kim, D.J.; Weilert, F.; Chan, H.L.Y.; Lalezari, J.; Hwang, S.G.; Nguyen, T.; Flores, O.; Hartman, G.; et al. Antiviral Activity, Safety, and Pharmacokinetics of Capsid Assembly Modulator NVR 3-778 in Patients with Chronic HBV Infection. Gastroenterology 2019, 156, 1392–1403.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, K.; Niu, J.; Ding, Y.; Gane, E.; Nguyen, T.; Alves, K.; Evanchick, M.; Zayed, H.; Huang, Q.; Knox, S.; et al. Antiviral activity, pharmacokinetics and safety of the secondgeneration hepatitis B core inhibitor ABI-H2158 in Phase 1b study of patients with HBeAg-positive chronic hepatitis B infection. J. Hepatol. 2020, 73 Suppl. 1, S125. [Google Scholar]
- BioSpace. Assembly Bio Announces Decision to Discontinue Clinical Development of ABI-H2158; Assembly Biosciences, Inc.: South San Francisco, CA, USA, 2021; Available online: https://www.biospace.com/article/releases/assembly-bio-announces-decision-to-discontinue-clinical-development-of-abi-h2158/ (accessed on 1 December 2021).
- Zoulim, F.; Yogaratnam, J.Z.; Vandenbossche, J.J.; Lenz, O.; Talloen, W.; Vistuer, C.; Moscalu, I.; Streinu-Cercel, A.; Bourgeois, S.; Buti, M.; et al. Safety, pharmacokinetics and antiviral activity of novel capsid assembly modulator (CAM) JNJ-56136379 (JNJ6379) in treatment-naïve chronic hepatitis B (CHB) patients without cirrhosis. J. Viral Hepat. 2018, 25, 36. [Google Scholar]
- Yuen, M.F.; Asselah, T.; Jacobson, I.M.; Brunetto, M.R.; Janssen, H.L.; Takehara, T.; Hou, J.L.; Kakuda, T.; Lambrecht, T.; Mauviel, M.B.; et al. Efficacy and safety of the siRNA JNJ-3989 and Capsid Assembly Modulator JNJ-6379 for the treatment of Chronic Hepatitis B virus infection(CHB): Results from the phase 2B Reef-1 study. In Proceedings of the an Oral Presentation at AASLD The Liver Meeting, Online, 15 November 2021. [Google Scholar]
- Gane, E.J.; Schwabe, C.; Lenz, O.; Verbinnen, T.; Talloen, W.; Kakuda, T.; Westland, C.; Patel, M.; Yogaratnam, J.; Van Remoortere, P. JNJ-64530440 (JNJ-0440), A Novel Class n Capsid Assembly Modulator (CAM-N): Safety, Tolerability, Pharmacokinetics (pk), and Antiviral Activity of Multiple Ascending Doses in Patients (PTS) with Chronic Hepatitis b (CHB). In Hepatology; WILEY: Hoboken, NJ, USA, 2019; Volume 70, p. 61A. [Google Scholar]
- Carey, I.; Gersch, J.; Wang, B.; Moigboi, C.; Kuhns, M.; Cloherty, G.; Dusheiko, G.; Agarwal, K. Pregenomic HBV RNA and Hepatitis B Core-Related Antigen Predict Outcomes in Hepatitis B e Antigen–Negative Chronic Hepatitis B Patients Suppressed on Nucleos(T)ide Analogue Therapy. Hepatology 2020, 72, 42–57. [Google Scholar] [CrossRef]
- Butler, E.K.; Gersch, J.; McNamara, A.; Luk, K.-C.; Holzmayer, V.; De Medina, M.; Schiff, E.; Kuhns, M.; Cloherty, G.A. Hepatitis B Virus Serum DNA andRNA Levels in Nucleos(t)ide Analog-Treated or Untreated Patients During Chronic and Acute Infection. Hepatology 2018, 68, 2106–2117. [Google Scholar] [CrossRef] [Green Version]
- Mak, L.-Y.; Wong, D.K.-H.; Cheung, K.-S.; Seto, W.-K.; Fung, J.; Yuen, M.-F. First-line oral antiviral therapies showed similar efficacies in suppression of serum HBcrAg in chronic hepatitis B patients. BMC Gastroenterol. 2021, 21, 1–7. [Google Scholar] [CrossRef]
- Noordeen, F.; Vaillant, A.; Jilbert, A.R. Nucleic Acid Polymers Inhibit Duck Hepatitis B Virus InfectionIn Vitro. Antimicrob. Agents Chemother. 2013, 57, 5291–5298. [Google Scholar] [CrossRef] [Green Version]
- Noordeen, F.; Vaillant, A.; Jilbert, A.R. Nucleic Acid Polymers Prevent the Establishment of Duck Hepatitis B Virus Infection In Vivo. Antimicrob. Agents Chemother. 2013, 57, 5299–5306. [Google Scholar] [CrossRef] [Green Version]
- Noordeen, F.; Scougall, C.A.; Grosse, A.; Qiao, Q.; Ajilian, B.B.; Reaiche-Miller, G.; Finnie, J.; Werner, M.; Broering, R.; Schlaak, J.F.; et al. Therapeutic Antiviral Effect of the Nucleic Acid Polymer REP 2055 against Persistent Duck Hepatitis B Virus Infection. PLoS ONE 2015, 10, e0140909. [Google Scholar] [CrossRef] [Green Version]
- Real, C.I.; Werner, M.; Paul, A.; Gerken, G.; Schlaak, J.F.; Vaillant, A.; Broering, R. Nucleic acid-based polymers effective against hepatitis B Virus infection in patients don’t harbor immunostimulatory properties in primary isolated liver cells. Sci. Rep. 2017, 7, srep43838. [Google Scholar] [CrossRef]
- Boulon, R.; Blanchet, M.; Vaillant, A.; Labonte, P. The hsp40 chaperone dnajb12 is involved in the morphogenesis of hbv spherical subviral particles and is selectively targeted by nucleic acid polymers. Hepatology 2020, 72, 53. [Google Scholar]
- Boulon, R.; Angélo, L.; Tétreault, Y.; Blanchet, M.; Vaillant, A.; Labonté, P. Ph-dependent interaction of NAPs with the HSP40 chaperone DNAJB12. In Proceedings of the HBV International Meeting, Toronto, Canada, 27 September 2021. [Google Scholar]
- Al-Mahtab, M.; Bazinet, M.; Vaillant, A. Safety and Efficacy of Nucleic Acid Polymers in Monotherapy and Combined with Immunotherapy in Treatment-Naive Bangladeshi Patients with HBeAg+ Chronic Hepatitis B Infection. PLoS ONE 2016, 11, e0156667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazinet, M.; Pântea, V.; Placinta, G.; Moscalu, I.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Iarovoi, L.; Smesnoi, V.; Musteata, T.; et al. Safety and Efficacy of 48 Weeks REP 2139 or REP 2165, Tenofovir Disoproxil, and Pegylated Interferon Alfa-2a in Patients with Chronic HBV Infection Naïve to Nucleos(t)ide Therapy. Gastroenterology 2020, 158, 2180–2194. [Google Scholar] [CrossRef]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Chen, Y.; Lu, M. Advances in Targeting the Innate and Adaptive Immune Systems to Cure Chronic Hepatitis B Virus Infection. Front. Immunol. 2020, 10, 3127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoletti, A.; Le Bert, N. Immunotherapy for Chronic Hepatitis B Virus Infection. Gut Liver 2018, 12, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broering, R.; Lu, M.; Schlaak, J.F. Role of Toll-like receptors in liver health and disease. Clin. Sci. 2011, 121, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Petes, C.; Odoardi, N.; Gee, K. The toll for trafficking: Toll-like receptor 7 delivery to the endosome. Front. Immunol. 2017, 4, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menne, S.; Tumas, D.B.; Liu, K.H.; Thampi, L.; AlDeghaither, D.; Baldwin, B.H.; Bellezza, C.A.; Cote, P.J.; Zheng, J.; Halcomb, R.; et al. Sustained efficacy and seroconversion with the Toll-like receptor 7 agonist GS-9620 in the Woodchuck model of chronic hepatitis B. J. Hepatol. 2015, 62, 1237–1245. [Google Scholar] [CrossRef] [Green Version]
- Gane, E.J.; Lim, Y.-S.; Gordon, S.C.; Visvanathan, K.; Sicard, E.; Fedorak, R.; Roberts, S.; Massetto, B.; Ye, Z.; Pflanz, S.; et al. The oral toll-like receptor-7 agonist GS-9620 in patients with chronic hepatitis B virus infection. J. Hepatol. 2015, 63, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Tan, A.T.; Ussher, J.; Sandalova, E.; Tang, X.-Z.; Tan-Garcia, A.; To, N.; Hong, M.; Chia, A.; Gill, U.S.; et al. Toll-Like Receptor 8 Agonist and Bacteria Trigger Potent Activation of Innate Immune Cells in Human Liver. PLoS Pathog. 2014, 10, e1004210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolowicz, K.E.; Iyer, R.P.; Czerwinski, S.; Suresh, M.; Yang, J.; Padmanabhan, S.; Sheri, A.; Pandey, R.K.; Skell, J.; Marquis, J.K.; et al. Antiviral Efficacy and Host Innate Immunity Associated with SB 9200 Treatment in the Woodchuck Model of Chronic Hepatitis B. PLoS ONE 2016, 11, e0161313. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühlbauer, M.; Fleck, M.; Schütz, C.; Weiss, T.; Froh, M.; Blank, C.; Schölmerich, J.; Hellerbrand, C. PD-L1 is induced in hepatocytes by viral infection and by interferon-α and-γ and mediates T cell apoptosis. J. Hepatol. 2006, 45, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral Intrahepatic T-Cell Responses Can Be Restored by Blocking Programmed Death-1 Pathway in Chronic Hepatitis B. Gastroenterology 2010, 138, 682–693.e4. [Google Scholar] [CrossRef]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of Hepatitis B Virus (HBV)-Specific T-Cell Dysfunction in Chronic HBV Infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef] [Green Version]
- Maier, H.; Isogawa, M.; Freeman, G.J.; Chisari, F.V. PD-1:PD-L1 Interactions Contribute to the Functional Suppression of Virus-Specific CD8+T Lymphocytes in the Liver. J. Immunol. 2007, 178, 2714–2720. [Google Scholar] [CrossRef] [Green Version]
- Kassel, R.; Cruise, M.W.; Iezzoni, J.C.; Taylor, N.A.; Pruett, T.L.; Hahn, Y.S. Chronically inflamed livers up-regulate expression of inhibitory B7 family members. Hepatology 2009, 50, 1625–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Shi, Y.; Li, S.; Zhang, Y.; Liu, Y.; Wu, Y.; Chen, Z. Blockade of T im-3 signaling restores the virus-specific CD 8+ T-cell response in patients with chronic hepatitis B. Eur. J. Immunol. 2012, 42, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Bertoletti, A.; Tanoto, T.A. PD-1/PD-L1 pathway and T-cell exhaustion in chronic hepatitis virus infection. J. Hepat. 2010, 17, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Qian, J.; Cui, Y.; Yan, Y.; He, H.; Wu, J.J. A Phase Iia Trial of Subcutaneously Administered Pd-L1 Antibody Asc22 (Envafolimab) In Patients with Chronic Hepatitis B. In Proceedings of the Oral Presentation at AASLD The Liver Meeting, Virtual, 12–15 November 2021. [Google Scholar]
- Huang, R.; Hao, Y.; Fan, Y.; Yang, C.; Wu, K.; Cao, S.; Wu, C. Association Between Cytotoxic T-Lymphocyte-Associated Antigen 4 +49A/G Polymorphism and Persistent Hepatitis B Virus Infection in the Asian Population: Evidence from the Current Studies. Genet. Test. Mol. Biomarkers 2013, 17, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kang, H.; Lee, H.H.; Kim, C.W. Programmed Cell Death 1 (PD-1) and Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4) in Viral Hepatitis. Int. J. Mol. Sci. 2017, 18, 1517. [Google Scholar] [CrossRef] [PubMed]
- Bohne, F.; Chmielewski, M.; Ebert, G.; Wiegmann, K.; Kürschner, T.; Schulze, A.; Urban, S.; Krönke, M.; Abken, H.; Protzer, U. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology 2008, 134, 239–247. [Google Scholar] [CrossRef]
- Gehring, A.; Xue, S.-A.; Ho, Z.Z.; Teoh, D.; Ruedl, C.; Chia, A.; Koh, S.; Lim, S.G.; Maini, M.; Stauss, H.; et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J. Hepatol. 2011, 55, 103–110. [Google Scholar] [CrossRef]
- Krebs, K.; Böttinger, N.; Huang, L.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T Cells Expressing a Chimeric Antigen Receptor That Binds Hepatitis B Virus Envelope Proteins Control Virus Replication in Mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Hoogeveen, R.C.; Boonstra, A. Checkpoint Inhibitors and Therapeutic Vaccines for the Treatment of Chronic HBV Infection. Front. Immunol. 2020, 11, 401. [Google Scholar] [CrossRef] [Green Version]
- Buchmann, P.; Dembek, C.; Kuklick, L.; Jäger, C.; Tedjokusumo, R.; von Freyend, M.J.; Drebber, U.; Janowicz, Z.; Melber, K.; Protzer, U. A novel therapeutic hepatitis B vaccine induces cellular and humoral immune responses and breaks tolerance in hepatitis B virus (HBV) transgenic mice. Vaccine 2013, 31, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Dubois, C.; Jacquier, E.; Dion, S.; Mancini-Bourgine, M.; Godon, O.; Kratzer, R.; Lelu-Santolaria, K.; Evlachev, A.; Meritet, J.-F.; et al. TG1050, an immunotherapeutic to treat chronic hepatitis B, induces robust T cells and exerts an antiviral effect in HBV-persistent mice. Gut 2015, 64, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Vandepapelière, P.; Lau, G.K.; Leroux-Roels, G.; Horsmans, Y.; Gane, E.; Tawandee, T.; bin Merican, M.I.; Win, K.M.; Trepo, C.; Cooksley, G.; et al. Therapeutic vaccination of chronic hepatitis B patients with virus suppression by antiviral therapy: A randomized, controlled study of co-administration of HBsAg/AS02 candidate vaccine and lamivudine. Vaccine 2007, 25, 8585–8597. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Blattman, J.N.; Ahmed, R. Low CD8 T-Cell Proliferative Potential and High Viral Load Limit the Effectiveness of Therapeutic Vaccination. J. Virol. 2005, 79, 8960–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Thompson, A.J.; Bowden, S.; Croagh, C.; Bell, S.; Desmond, P.V.; Levy, M.; Locarnini, S.A. Hepatitis B surface antigen levels during the natural history of chronic hepatitis B: A perspective on Asia. J. Hepatol. 2010, 52, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, E.; Ma, Z.; Wu, W.; Kosinska, A.; Zhang, X.; Möller, I.; Seiz, P.; Glebe, D.; Wang, B.; et al. Enhancing virus-specific immunity in vivo by combining therapeutic vaccination and PD-L1 blockade in chronic hepadnaviral infection. PLoS Pathog. 2014, 10, e1003856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, S.-J.; Mueller, S.; Wherry, E.J.; Barber, D.L.; Aubert, R.D.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Enhancing therapeutic vaccination by blocking PD-1–mediated inhibitory signals during chronic infection. J. Exp. Med. 2008, 205, 543–555. [Google Scholar] [CrossRef] [Green Version]
- Benechet, A.; De Simone, G.; Di Lucia, P.; Cilenti, F.; Barbiera, G.; Le Bert, N.; Fumagalli, V.; Lusito, E.; Moalli, F.; Bianchessi, V.; et al. Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nature 2019, 574, 200–205. [Google Scholar] [CrossRef]
- Gane, E.; Verdon, D.J.; Brooks, A.E.; Gaggar, A.; Nguyen, A.H.; Subramanian, G.M.; Schwabe, C.; Dunbar, R. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J. Hepatol. 2019, 71, 900–907. [Google Scholar] [CrossRef]
- Yan, H.; Liu, Y.; Sui, J.; Li, W. NTCP opens the door for hepatitis B virus infection. Antivir. Res. 2015, 121, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Bonino, F.; Heermann, K.H.; Rizzetto, M.; Gerlich, W.H. Hepatitis delta virus: Protein composition of delta antigen and its hepatitis B virus-derived envelope. J. Virol. 1986, 58, 945–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis δ virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef] [Green Version]
- Rizzetto, M.; Alavian, S.M. Hepatitis delta. The rediscovery. Clin. Liver Dis. 2013, 17, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Liver EAFTSOT. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedemeyer, H.; Yurdaydìn, C.; Dalekos, G.N.; Erhardt, A.; Çakaloğlu, Y.; Değertekin, H.; Gürel, S.; Zeuzem, S.; Zachou, K.; Bozkaya, H.; et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N. Engl. J. Med. 2011, 364, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedemeyer, H.; Yurdaydin, C.; Hardtke, S.; Caruntu, F.A.; Curescu, M.G.; Yalcin, K.; Akarca, U.S.; Gürel, S.; Zeuzem, S.; Erhardt, A.; et al. Peginterferon alfa-2a plus tenofovir disoproxil fumarate for hepatitis D (HIDIT-II): A randomised, placebo controlled, phase 2 trial. Lancet Infect. Dis. 2019, 19, 275–286. [Google Scholar] [CrossRef]
- Heller, T.; Rotman, Y.; Koh, C.; Clark, S.; Haynes-Williams, V.; Chang, R.; McBurney, R.; Schmid, P.; Albrecht, J.; Kleiner, D.E.; et al. Long-term therapy of chronic delta hepatitis with peginterferon alfa. Aliment. Pharmacol. Ther. 2014, 40, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-Lambda (IFN-λ) Is Expressed in a Tissue-Dependent Fashion and Primarily Acts on Epithelial Cells In Vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef]
- Etzion, O.; Hamid, S.S.; Lurie, Y.; Gane, E.; Bader, N.; Yardeni, D.; Nevo-Shor, A.; Channa, S.; Mawani, M.; Parkash, O.; et al. PS-052-End of study results from LIMT HDV study: 36% durable virologic response at 24 weeks post-treatment with pegylated interferon lambda monotherapy in patients with chronic hepatitis delta virus infection. J. Hepatol. 2019, 70, e32. [Google Scholar] [CrossRef]
- Koh, C.; Hercun, J.; Rahman, F.; Huang, A.; Da, B.; Surana, P.; Kapuria, D.; Rotman, Y.; Vittal, A.; Gilman, C.A.; et al. A Phase 2 Study of Peginterferon Lambda, Lonafarnib and Ritonavir for 24 Weeks: End-of-Treatment Results from the LIFT HDV Study. J. Hepatol. 2020, 73, S130. [Google Scholar] [CrossRef]
- Liu, M.; Bryant, M.S.; Chen, J.; Lee, S.; Yaremko, B.; Lipari, P.; Malkowski, M.; Ferrari, E.; Nielsen, L.; Prioli, N.; et al. Antitumor activity of SCH 66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and wap-ras transgenic mice. Cancer Res. 1998, 58, 4947–4956. [Google Scholar] [PubMed]
- Bordier, B.B.; Ohkanda, J.; Liu, P.; Lee, S.Y.; Salazar, F.H.; Marion, P.L.; Ohashi, K.; Meuse, L.; Kay, M.A.; Casey, J.L.; et al. In vivo antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J. Clin. Investig. 2003, 112, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, C.; Canini, L.; Dahari, H.; Zhao, X.; Uprichard, S.L.; Haynes-Williams, V.; Winters, M.A.; Subramanya, G.; Cooper, S.L.; Pinto, P.; et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: A proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect. Dis. 2015, 15, 1167–1174. [Google Scholar] [CrossRef] [Green Version]
- Urdaydin, C.; Keskin, O.; Kalkan, Ç.; Karakaya, F.; Çalişkan, A.; Karatayli, E.; Karatayli, S.; Bozdayi, A.M.; Koh, C.; Heller, T.; et al. Optimizing lonafarnib treatment for the management of chronic delta hepatitis: The LOWR HDV-1 study. Hepatology 2018, 67, 1224–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurdaydin, C.; Idilman, R.; Keskin, O.; Kalkan, C.; Karakaya, M.; Caliskan, A.; Yurdcu, E.; Karatayli, S.; Bozdayi, M.; Koh, C.; et al. A phase 2 dose-optimization study of lonafarnib with ritonavir for the treatment of chronic delta hepatitis—End of treatment results from the LOWR HDV-2 study. J. Hepatol. 2017, 66, S33–S34. [Google Scholar] [CrossRef]
- Koh, C.; Surana, P.; Han, T.; Fryzek, N.; Kapuria, D.; Etzion, O.; Takyar, V.; Rotman, Y.; Canales, R.; Dahari, H.; et al. A phase 2 study exploring once daily dosing of ritonavir boosted lonafarnib for the treatment of chronic delta hepatitis—End of study results from the LOWR HDV-3 study. J. Hepatol. 2017, 66, S101–S102. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Port, K.; Deterding, K.; Wranke, A.; Kirschner, J.; Bruno, B.; Martins, B.; Glenn, J.; Kornberg, M.; Manns, M. A phase 2 dose-escalation study of lonafarnib plus ritonavir in patients with chronic hepatitis D: Final results from the Lonafarnib with ritonavir in HDV-4 (LOWR HDV-4) study. J. Hepatol. 2017, 66, S24. [Google Scholar] [CrossRef]
- Koh, C.; Da, B.L.; Surana, P.; Huang, A.; Kapuria, D.; Rotman, Y.; Vittal, A.; Gilman, C.; Ben-Yakov, G.; Lai, C.; et al. A Phase 2 Study of Lonafarnib, Ritonavir and Peginterferon Lambda for 24 Weeks: Interim End-of-Treatment Results from the Lift Hdv Study. In Hepatology; WILEY: Hoboken, NJ, USA, 2019; Volume 70, p. 1483A. [Google Scholar]
- Kang, C.; Syed, Y.Y. Bulevirtide: First Approval. Drugs 2020, 80, 1601–1605. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Bogomolov, P.; Blank, A.; Allweiss, L.; Dandri-Petersen, M.; Bremer, B.; Voronkova, N.; Schöneweis, K.; Pathil, A.; Burhenne, J.; et al. Final results of a multicenter, open-label phase 2b clinical trial to assess safety and efficacy of Myrcludex B in combination with Tenofovir in patients with chronic HBV/HDV co-infection. J. Hepatol. 2018, 68, S3. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Schöneweis, K.; Bogomolov, P.O.; Voronkova, N.; Chulanov, V.; Stepanova, T.; Bremer, B.; Allweiss, L.; Dandri, M.; Burhenne, J.; et al. GS-13-Final results of a multicenter, open-label phase 2 clinical trial (MYR203) to assess safety and efficacy of myrcludex B in cwith PEG-interferon Alpha 2a in patients with chronic HBV/HDV co-infection. J. Hepatol. 2019, 70, e81. [Google Scholar] [CrossRef]
- Bazinet, M.; Pântea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Albrecht, J.; Schmid, P.; Le Gal, F.; Gordien, E.; Krawczyk, A.; et al. Safety and efficacy of REP 2139 and pegylated interferon alfa-2a for treatment-naive patients with chronic hepatitis B virus and hepatitis D virus co-infection (REP 301 and REP 301-LTF): A non-randomised, open-label, phase 2 trial. Lancet Gastroenterol. Hepatol. 2017, 2, 877–889. [Google Scholar] [CrossRef]
- Bazinet, M.; Pântea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Anderson, M.; Gersch, J.; Holzmayer, V.; Elsner, C.; Krawczyk, A.; et al. Persistent Control of Hepatitis B Virus and Hepatitis Delta Virus Infection Following REP 2139-Ca and Pegylated Interferon Therapy in Chronic Hepatitis B Virus/Hepatitis Delta Virus Coinfection. Hepatol. Commun. 2021, 5, 189–202. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Partial Cure | Functional Cure | Complete Cure |
|---|---|---|
| HBV DNA low or not detected | HBV DNA not detected | HBV DNA not detected |
| HBeAg negative | HBeAg negative | HBeAg negative |
| HBsAg positive | HBsAg negative ± anti-HBs | HBsAg negative ± anti-HBs |
| cccDNA detected | cccDNA detected but not active | cccDNA not detected |
| Integrated HBV DNA detected | Integrated HBV DNA detected | Integrated HBV DNA not detected |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, I.W.; Dad Ullah, M.U.; Choudhry, M.; Ali, M.J.; Ali, M.A.; Lam, S.L.K.; Shah, P.A.; Kaur, S.P.; Lau, D.T.Y. Novel Therapies of Hepatitis B and D. Microorganisms 2021, 9, 2607. https://doi.org/10.3390/microorganisms9122607
Khan IW, Dad Ullah MU, Choudhry M, Ali MJ, Ali MA, Lam SLK, Shah PA, Kaur SP, Lau DTY. Novel Therapies of Hepatitis B and D. Microorganisms. 2021; 9(12):2607. https://doi.org/10.3390/microorganisms9122607
Chicago/Turabian StyleKhan, Iman Waheed, Mati Ullah Dad Ullah, Mina Choudhry, Mukarram Jamat Ali, Muhammad Ashar Ali, Sam L. K. Lam, Pir Ahmad Shah, Satinder Pal Kaur, and Daryl T. Y. Lau. 2021. "Novel Therapies of Hepatitis B and D" Microorganisms 9, no. 12: 2607. https://doi.org/10.3390/microorganisms9122607
APA StyleKhan, I. W., Dad Ullah, M. U., Choudhry, M., Ali, M. J., Ali, M. A., Lam, S. L. K., Shah, P. A., Kaur, S. P., & Lau, D. T. Y. (2021). Novel Therapies of Hepatitis B and D. Microorganisms, 9(12), 2607. https://doi.org/10.3390/microorganisms9122607