
Comparative Analysis of Salmon Cell Lines and Zebrafish Primary Cell Cultures Infection with the Fish Pathogen Piscirickettsia salmonis


 and
and
Abstract
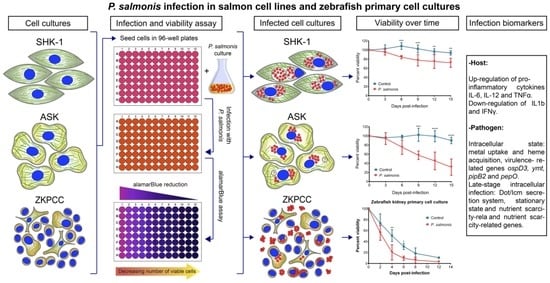
:
1. Introduction
2. Materials and Methods
2.1. Bacteria, Media, and Growth Conditions
2.2. Infection and Culture Conditions for Salmon-Derived Cell Lines
2.3. Zebrafish Husbandry and Generation of Zebrafish Primary Cell Cultures
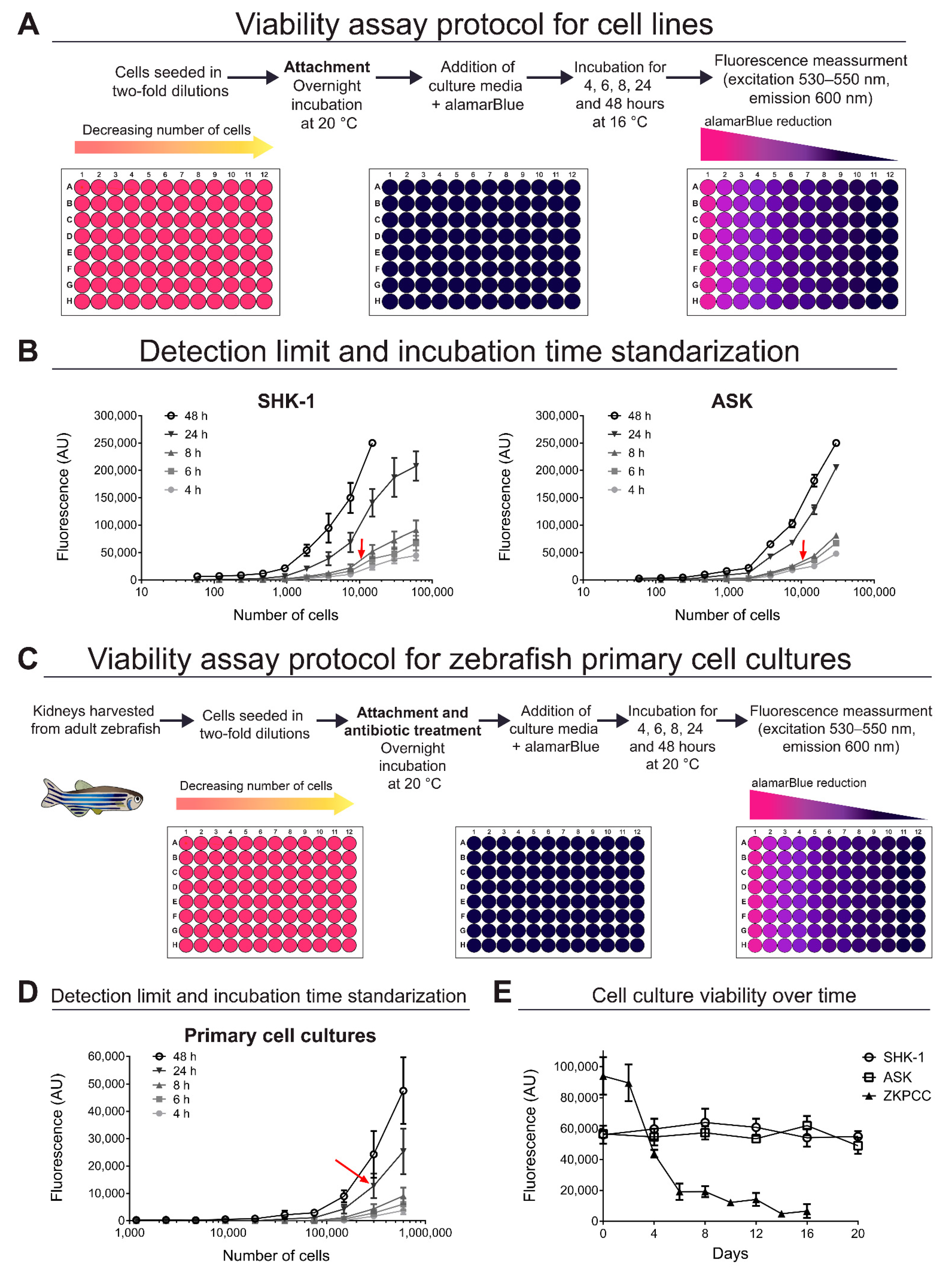
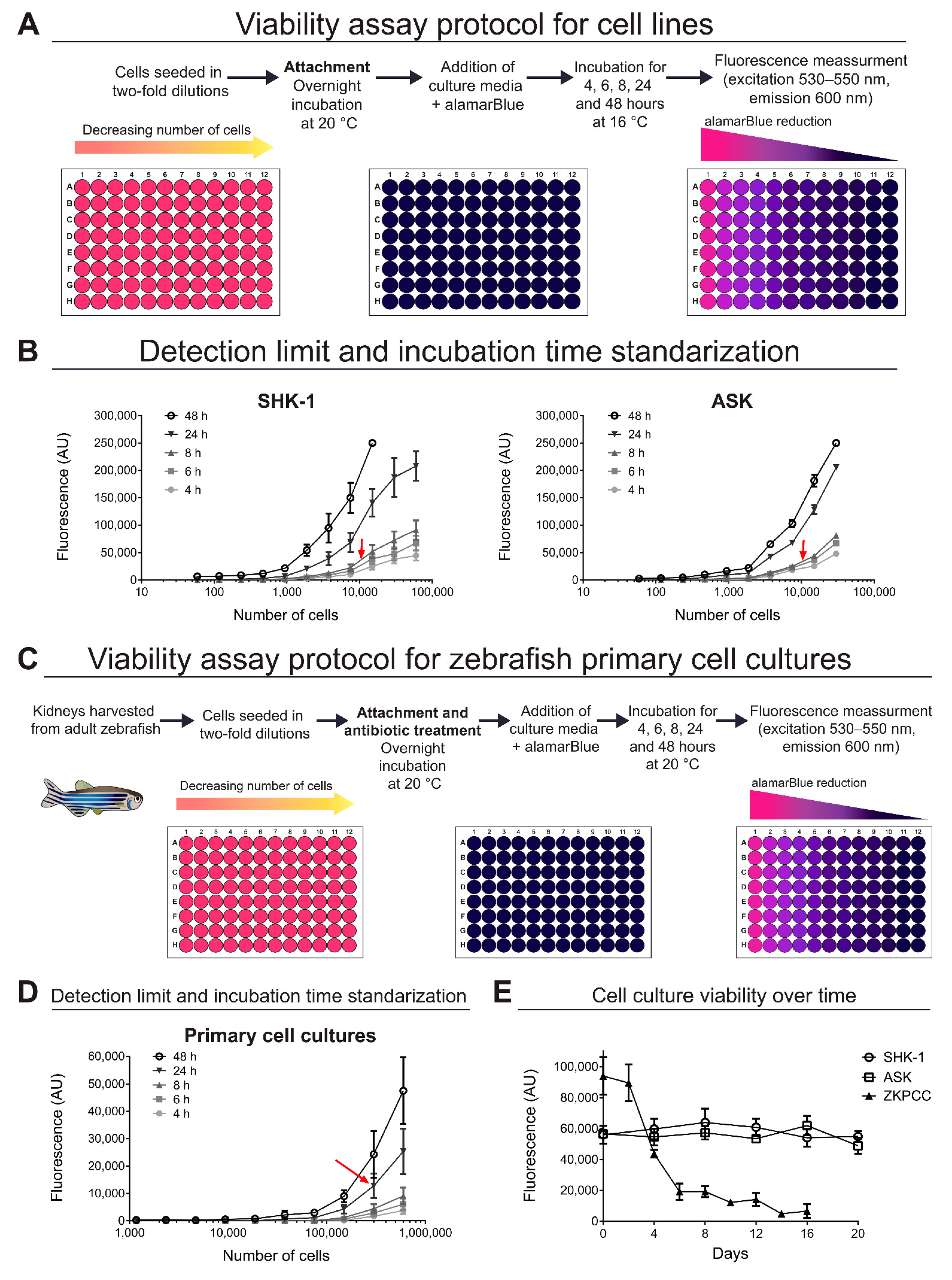
2.4. Standardization of Cell Viability Assays
2.5. Viability Assays in Infected Cell Cultures
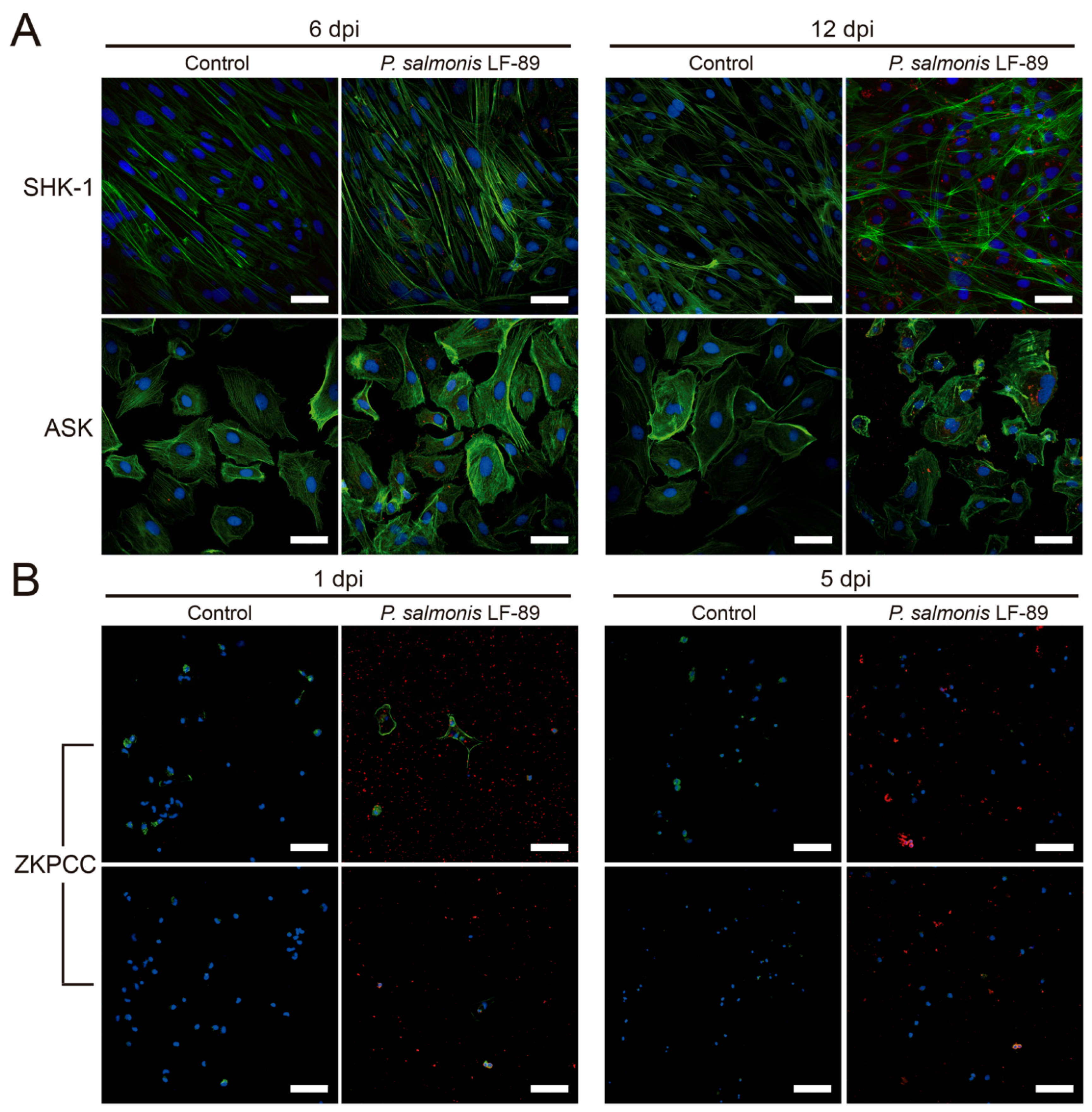
2.6. Immunofluorescence Microscopy and Antibody Staining
2.7. RNA Extraction from P. salmonis and Cell Cultures
2.8. Quantitative Real-Time PCR Assays (qPCR)
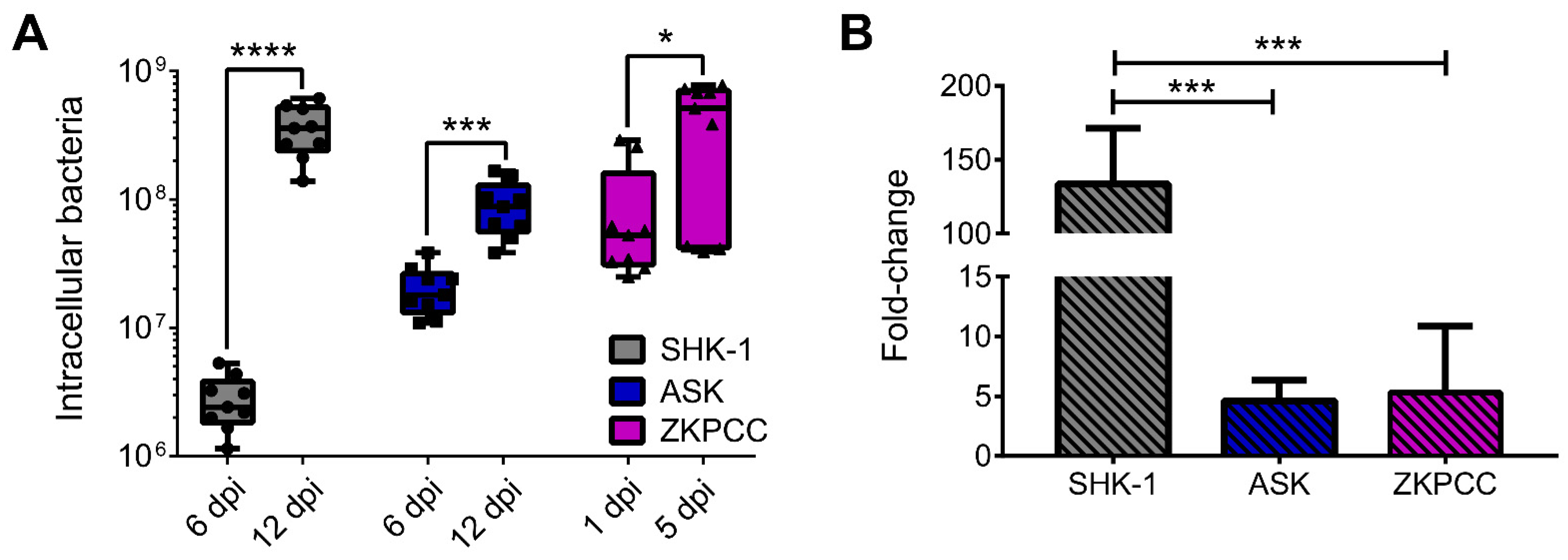
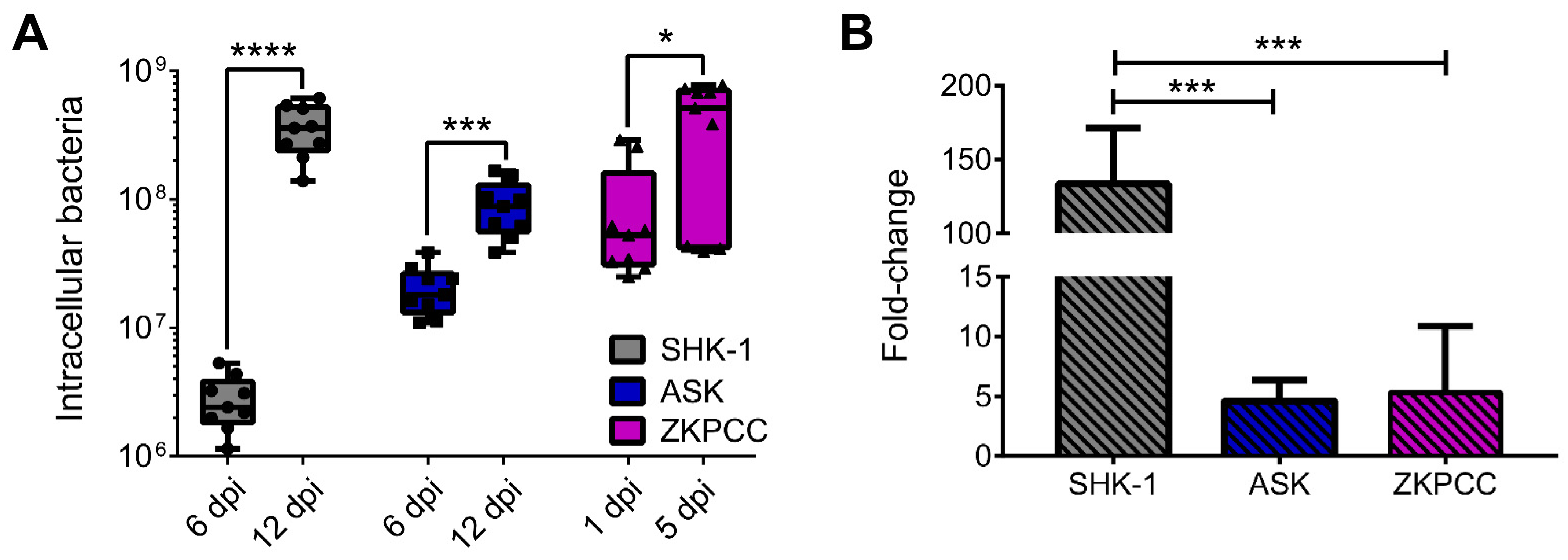
2.9. P. salmonis Quantification inside Cell Cultures
2.10. Statistical Analysis
3. Results and Discussions
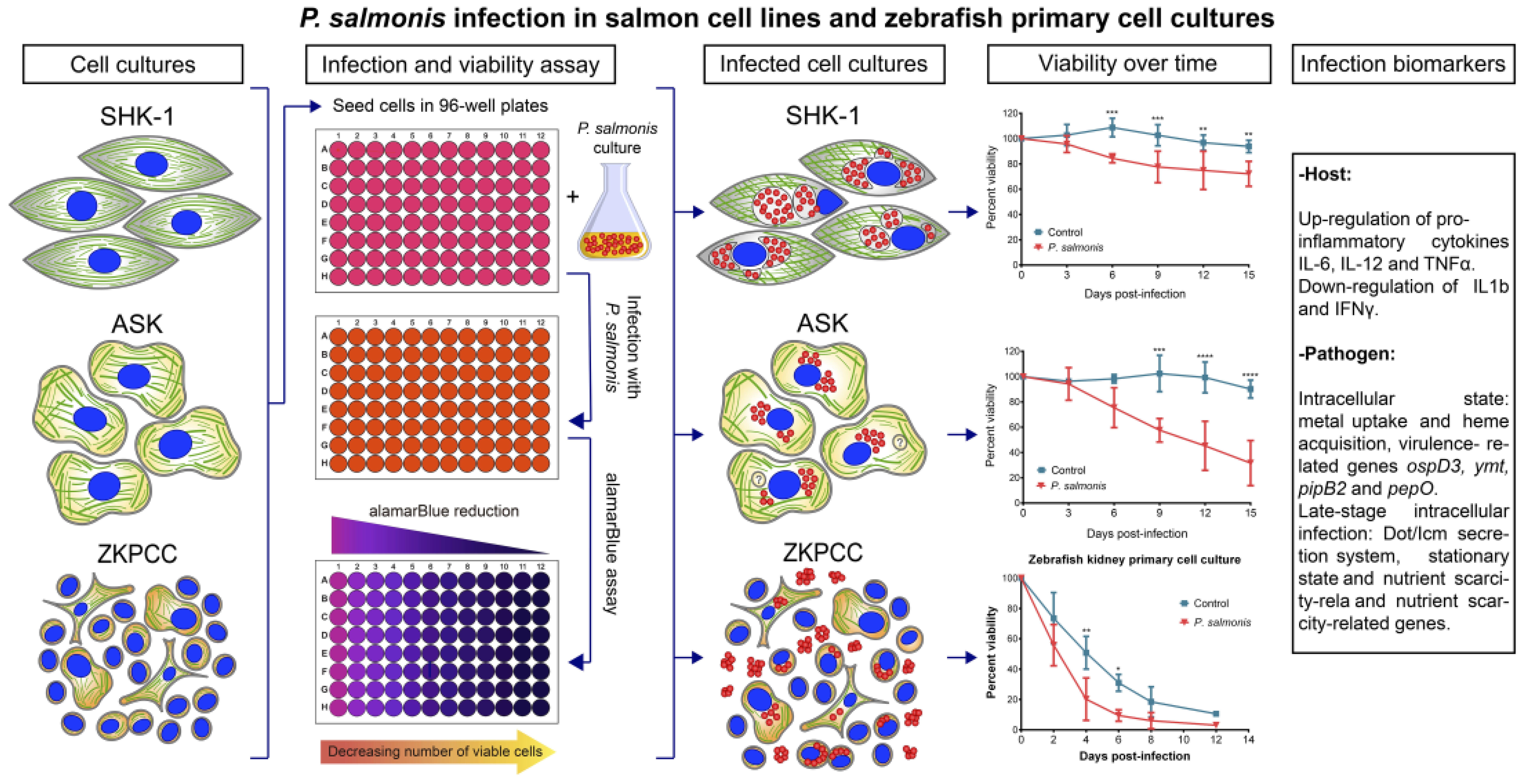
3.1. Viability of Cell Cultures over Time
3.2. Phenotypic Effects of P. salmonis Infection in Cell Cultures
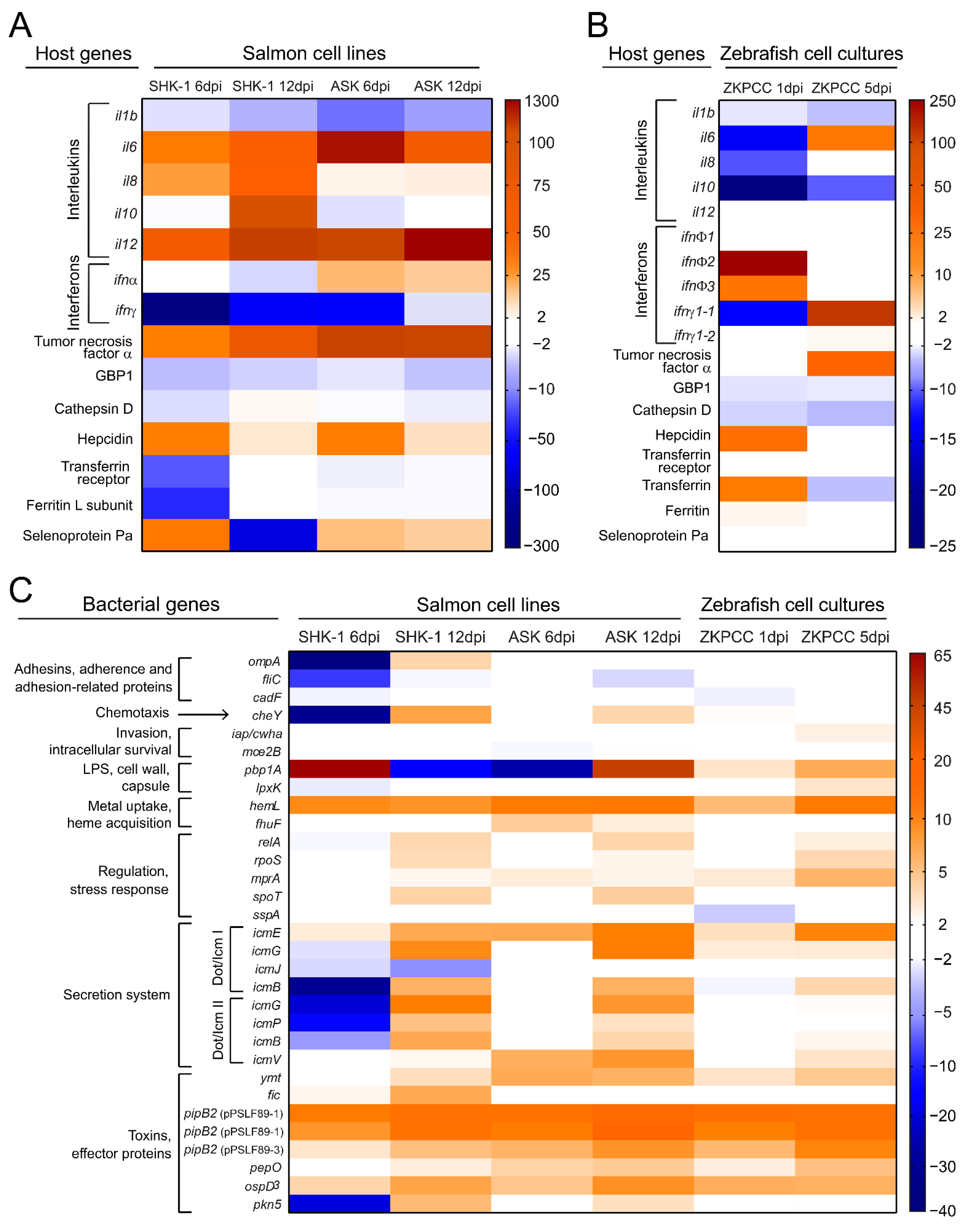
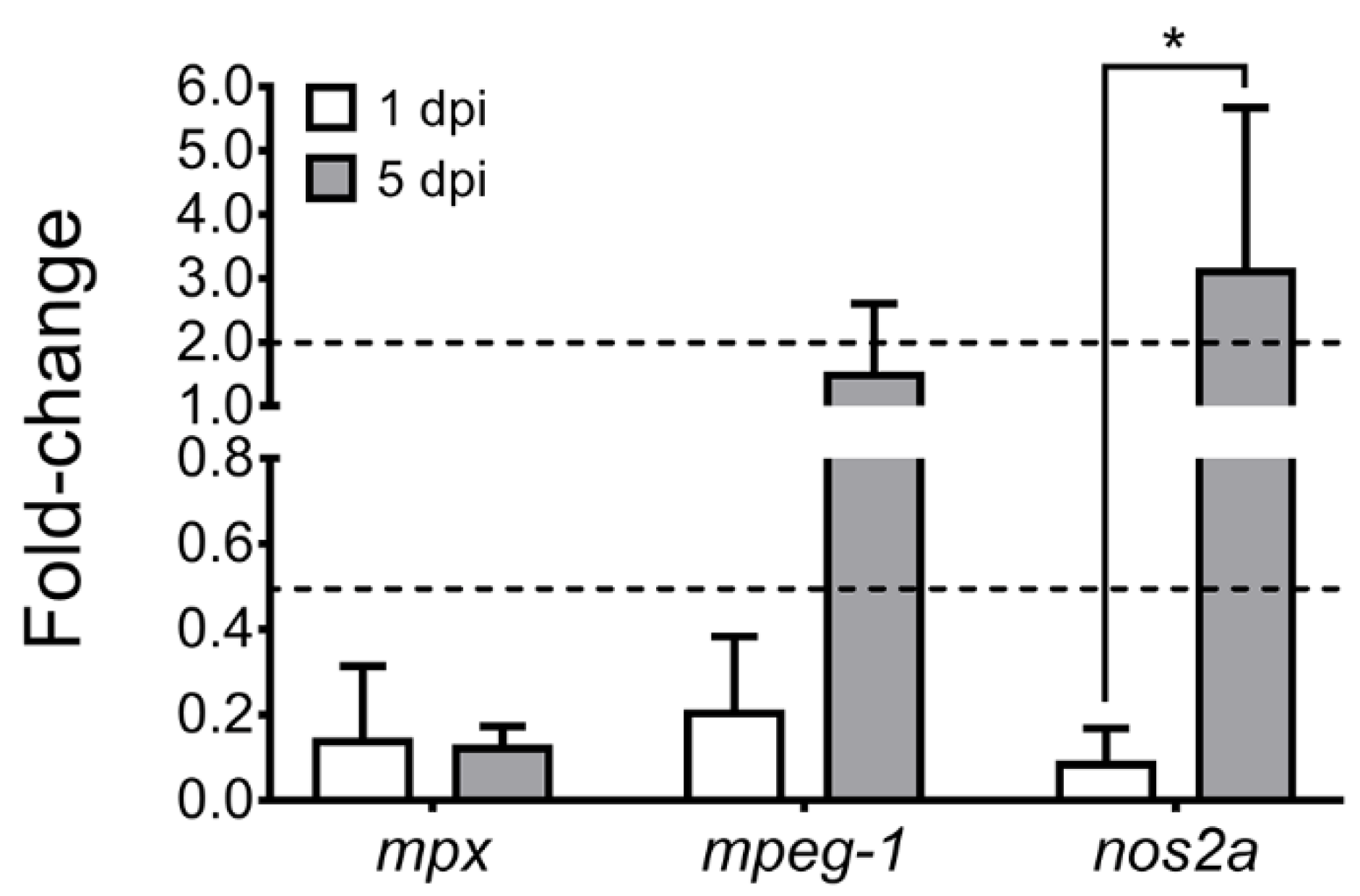
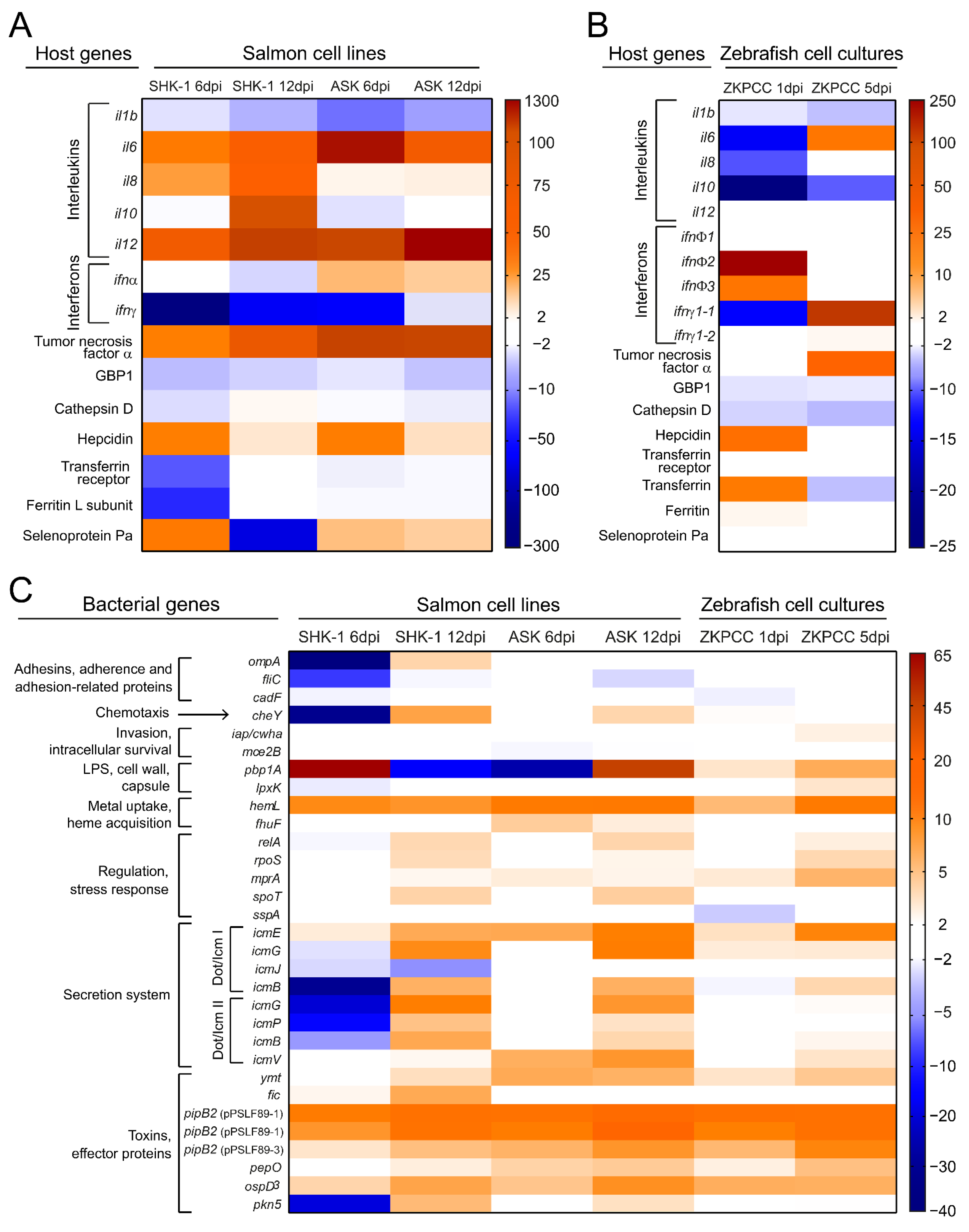
3.3. Host Immune Response and P. salmonis Virulence Factors during Infection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fryer, J.L.; Lannan, C.N.; Giovannoni, S.J.; Wood, N.D. Piscirickettsia salmonis gen. nov., sp. nov., the causative agent of an epizootic disease in salmonid fishes. Int. J. Syst. Bacteriol. 1992, 42, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Mauel, M.J.; Giovannoni, S.J.; Fryer, J.L. Phylogenetic analysis of Piscirickettsia salmonis by 16S, internal transcribed spacer (ITS) and 23S ribosomal DNA sequencing. Dis. Aquat. Organ. 1999, 35, 115–123. [Google Scholar] [CrossRef]
- Almendras, F.E.; Fuentealba, I.C. Salmonid rickettsial septicemia caused by Piscirickettsia salmonis: A review. Dis. Aquat. Organ. 1997, 29, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Rozas, M.; Enríquez, R.; Enr, R. Piscirickettsiosis and Piscirickettsia salmonis in fish: A review. J. Fish Dis. 2014, 37, 163–188. [Google Scholar] [CrossRef]
- Mauel, M.J.; Miller, D.L. Piscirickettsiosis and piscirickettsiosis-like infections in fish: A review. Vet. Microbiol. 2002, 87, 279–289. [Google Scholar] [CrossRef]
- Bravo, S.; Campos, M. Coho salmon syndrome in Chile. Am. Fish. Soc. Newsl. 1989, 17, 3. [Google Scholar]
- Cvitanich, J.D.D.; Garate, N.O.; Smith, C.E.E.; Garate, O.; Smith, C.E.E. The isolation of a rickettsia-like organism causing disease and mortality in Chilean salmonids and its confirmation by Koch’s postulate. J. Fish Dis. 1991, 14, 121–145. [Google Scholar] [CrossRef]
- Fryer, J.L.; Lannan, C.N.; Garces, L.H.; Larenas, J.J.; Smith, P.A. Isolation of a rickettsiales-like organism from diseased coho salmon (Oncorhynchus kisutch) in Chile. Fish Pathol. 1990, 25, 107–114. [Google Scholar] [CrossRef]
- McCarthy, U.; Steiropoulos, N.A.; Thompson, K.D.; Adams, A.; Ellis, A.E.; Ferguson, H.W. Confirmation of Piscirickettsia salmonis as a pathogen in European sea bass Dicentrarchus labrax and phylogenetic comparison with salmonid strains. Dis. Aquat. Organ. 2005, 64, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.F.; Yun, S.; Marty, G.D.; McDowell, T.S.; House, M.L.; Appersen, J.A.; Guenther, T.A.; Arkush, K.D.; Hedrick, R.P. A Piscirickettsia salmonis-like bacterium associated with mortality of white seabass Atractoscion nobilis. Dis. Aquat. Organ. 2000, 43, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Mauel, M.J.; Miller, D.L.; Frazier, K.; Liggett, A.D.; Styer, L.; Montgomery-Brock, D.; Brock, J. Characterization of a piscirickettsiosis-like disease in Hawaiian tilapia. Dis. Aquat. Organ. 2003, 53, 249–255. [Google Scholar] [CrossRef]
- Contreras-Lynch, S.; Olmos, P.; Vargas, A.; Figueroa, J.; González-Stegmaier, R.; Enríquez, R.; Romero, A. Identification and genetic characterization of Piscirickettsia salmonis in native fish from southern Chile. Dis. Aquat. Organ. 2015, 115, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Departamento de Salud Animal; Subdirección de Acuicultura; de Acuicultura, S.N.P.Y. Informe Sanitario de la Salmonicultura en Centros Marinos año 2020; Chilean National Fisheries and Aquaculture Service: Valparaíso, Chile, 2021.
- Sernapesca Informe Sobre uso de Antimicrobianos en la Salmonicultura Nacional-año 2019; Chilean National Fisheries and Aquaculture Service: Valparaíso, Chile, 2020.
- Smith, P.R.; Le Breton, A.; Horberg, T.E.; Corsin, F. Guidelines for antimicrobial use in aquaculture. In Guide to Antimicrobial Use in Animals; Guardabassi, L., Jensen, L.B., Kruse, H., Eds.; Blackwell Publishing: Oxford, UK, 2009; pp. 207–218. [Google Scholar]
- Miranda, C.D.; Godoy, F.A.; Lee, M.R. Current status of the use of antibiotics and the antimicrobial resistance in the chilean salmon farms. Front. Microbiol. 2018, 9, 1284. [Google Scholar] [CrossRef] [PubMed]
- Cabello, F.C.; Godfrey, H.P.; Buschmann, A.H.; Dölz, H.J. Aquaculture as yet another environmental gateway to the development and globalisation of antimicrobial resistance. Lancet Infect. Dis. 2016, 16, e127–e133. [Google Scholar] [CrossRef]
- Díaz, S.; Rojas, M.E.; Galleguillos, M.; Maturana, C.; Smith, P.I.; Cifuentes, F.; Contreras, I.; Smith, P.A. Apoptosis inhibition of Atlantic salmon (Salmo salar) peritoneal macrophages by Piscirickettsia salmonis. J. Fish Dis. 2017, 40, 1895–1902. [Google Scholar] [CrossRef]
- Yañez, A.J.; Valenzuela, K.; Silva, H.; Retamales, J.; Romero, A.; Enriquez, R.; Figueroa, J.; Claude, A.; Gonzalez, J.; Carcamo, J.G. Broth medium for the successful culture of the fish pathogen Piscirickettsia salmonis. Dis. Aquat. Organ. 2012, 97, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Rojas, V.; Galanti, N.; Bols, N.C.; Marshall, S.H. Productive infection of Piscirickettsia salmonis in macrophages and monocyte-like cells from rainbow trout, a possible survival strategy. J. Cell. Biochem. 2009, 108, 631–637. [Google Scholar] [CrossRef]
- Tandberg, J.; Oliver, C.; Lagos, L.; Gaarder, M.; Yáñez, A.J.; Ropstad, E.; Winther-Larsen, H.C. Membrane vesicles from Piscirickettsia salmonis induce protective immunity and reduce development of salmonid rickettsial septicemia in an adult zebrafish model. Fish Shellfish Immunol. 2017, 67, 189–198. [Google Scholar] [CrossRef]
- Tandberg, J.I.; Lagos, L.X.; Langlete, P.; Berger, E.; Rishovd, A.L.; Roos, N.; Varkey, D.; Paulsen, I.T.; Winther-Larsen, H.C. Comparative analysis of membrane vesicles from three Piscirickettsia salmonis isolates reveals differences in vesicle characteristics. PLoS ONE 2016, 11, e0165099. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, A.; Aravena, P.; Pulgar, R.; Travisany, D.; Ortiz-Severín, J.; Chávez, F.P.F.P.; Maass, A.; González, M.; Cambiazo, V. Transcriptomic changes of Piscirickettsia salmonis during intracellular growth in a salmon macrophage-like cell line. Front. Cell. Infect. Microbiol. 2020, 9, 426. [Google Scholar] [CrossRef]
- Verónica Rojas, M.; Olivares, P.J.; del Río, R.; Marshall, S.H. Characterization of a novel and genetically different small infective variant of Piscirickettsia salmonis. Microb. Pathog. 2008, 44, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Yañez, A.J.; Silva, H.; Valenzuela, K.; Pontigo, J.P.; Godoy, M.; Troncoso, J.; Romero, A.; Figueroa, J.; Carcamo, J.G.; Avendaño-Herrera, R. Two novel blood-free solid media for the culture of the salmonid pathogen Piscirickettsia salmonis. J. Fish Dis. 2013, 36, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Severín, J.; Travisany, D.; Maass, A.; Cambiazo, V.; Chávez, F.P. Global proteomic profiling of Piscirickettsia salmonis and salmon macrophage-like cells during intracellular infection. Microorganisms 2020, 8, 1845. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, C.B.; Ostoji, J.; Tell, R.M.; Roth, J.A. Zebrafish (Danio rerio) whole kidney assays to measure neutrophil extracellular trap release and degranulation of primary granules. J. Immunol. Methods 2007, 319, 87–97. [Google Scholar]
- Czekanska, E.M. Assesment of cell proliferation with resazurin-based fluorescent dye. In Mammalian Cell Viability; Clifton, N.J., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; Volume 740, pp. 27–32. ISBN 978-1-61779-107-9. [Google Scholar]
- Pagé, B.; Pagé, M.; Noel, C. A new fluorometric assay for cytotoxic measurements in vitro. Int. J. Oncol. 1993, 3, 473–476. [Google Scholar]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Ramakers, C.; Ruijter, J.M.; Lekanne Deprez, R.H.; Moorman, A.F.M. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]
- Schreer, A.; Tinson, C.; Sherry, J.P.; Schirmer, K. Application of Alamar blue/5-carboxyfluorescein diacetate acetoxymethyl ester as a noninvasive cell viability assay in primary hepatocytes from rainbow trout. Anal. Biochem. 2005, 344, 76–85. [Google Scholar] [CrossRef]
- Hamalainen-Laanaya, H.K.; Orloff, M.S. Analysis of cell viability using time-dependent increase in fluorescence intensity. Anal. Biochem. 2012, 429, 32–38. [Google Scholar] [CrossRef]
- Mo, C.; Yamagata, R.; Pan, A.; Reddy, J.; Hazari, N.; Duke, G. Development of a high-throughput Alamar blue assay for the determination of influenza virus infectious dose, serum antivirus neutralization titer and virus ca/ts phenotype. J. Virol. Methods 2008, 150, 63–69. [Google Scholar] [CrossRef]
- Stachura, D.L.; Reyes, J.R.; Bartunek, P.; Paw, B.H.; Zon, L.I.; Traver, D. Zebrafish kidney stromal cell lines support multilineage hematopoiesis. Blood 2009, 114, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.A.; Díaz, F.E.; Rojas, M.E.; Díaz, S.; Galleguillos, M.; Carbonero, A. Effect of Piscirickettsia salmonis inoculation on the ASK continuous cell line. J. Fish Dis. 2015, 38, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Isla, A.; Haussmann, D.; Vera, T.; Kausel, G.; Figueroa, J. Identification of the clpB and bipA genes and an evaluation of their expression as related to intracellular survival for the bacterial pathogen Piscirickettsia salmonis. Vet. Microbiol. 2014, 173, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Traver, D.; Paw, B.H.; Poss, K.D.; Penberthy, W.T.; Lin, S.; Zon, L.I. Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol. 2003, 4, 1238–1246. [Google Scholar] [CrossRef]
- Hall, C.J.; Flores, M.V.; Oehlers, S.H.; Sanderson, L.E.; Lam, E.Y.; Crosier, K.E.; Crosier, P.S. Infection-responsive expansion of the hematopoietic stem and progenitor cell compartment in zebrafish is dependent upon inducible nitric oxide. Cell Stem Cell 2012, 10, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Rise, M.L.M.; Jones, S.S.R.M.; Brown, G.D.; von Schalburg, K.R.; Davidson, W.S.; Koop, B.F. Microarray analyses identify molecular biomarkers of Atlantic salmon macrophage and hematopoietic kidney response to Piscirickettsia salmonis infection. Physiol. Genom. 2004, 20, 21–35. [Google Scholar] [CrossRef]
- Rozas-Serri, M.; Peña, A.; Maldonado, L. Transcriptomic profiles of post-smolt Atlantic salmon challenged with Piscirickettsia salmonis reveal a strategy to evade the adaptive immune response and modify cell-autonomous immunity. Dev. Comp. Immunol. 2018, 81, 348–362. [Google Scholar] [CrossRef]
- Pulgar, R.; Hödar, C.; Travisany, D.; Zuñiga, A.; Domínguez, C.; Maass, A.; González, M.; Cambiazo, V. Transcriptional response of Atlantic salmon families to Piscirickettsia salmonis infection highlights the relevance of the iron-deprivation defence system. BMC Genom. 2015, 16, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacchi, L.; Bron, J.E.; Taggart, J.B.; Secombes, C.J.; Bickerdike, R.; Adler, M.A.; Takle, H.; Martin, S.A.M. Multiple tissue transcriptomic responses to Piscirickettsia salmonis in Atlantic salmon (Salmo salar). Physiol Genom. 2011, 43, 1241–1254. [Google Scholar] [CrossRef] [Green Version]
- Salazar, C.; Haussmann, D.; Kausel, G.; Figueroa, J. Molecular cloning of Salmo salar Toll-like receptors (TLR1, TLR22, TLR5M and TLR5S) and expression analysis in SHK-1 cells during Piscirickettsia salmonis infection. J. Fish Dis. 2015, 39, 239–248. [Google Scholar] [CrossRef]
- Alvarez, C.A.; Gomez, F.A.; Mercado, L.; Ramirez, R.; Marshall, S.H. Piscirickettsia salmonis imbalances the innate immune response to succeed in a productive infection in a salmonid cell line model. PLoS ONE 2016, 11, e0163943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieger, D.; Stein, C.; Neifer, D.; Van Der Sar, A.M.; Leptin, M. The role of gamma interferon in innate immunity in the zebrafish embryo. DMM Dis. Model. Mech. 2009, 2, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Aggad, D.; Mazel, M.; Boudinot, P.; Mogensen, K.E.; Hamming, O.J.; Hartmann, R.; Kotenko, S.; Herbomel, P.; Lutfalla, G.; Levraud, J.-P. The Two Groups of Zebrafish Virus-Induced Interferons Signal via Distinct Receptors with Specific and Shared Chains. J. Immunol. 2009, 183, 3924–3931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parhi, J.; Sahoo, L.; Choudhury, J.; Choudhury, T.G.; Baruah, A.; Paniprasad, K.; Makesh, M. Molecular characterization and expression analysis of interferon γ (IFN-γ) gene in Labeo rohita (Ham.). Aquac. Rep. 2015, 2, 97–105. [Google Scholar] [CrossRef]
- Valenzuela-Miranda, D.; Gallardo-Escárate, C. Novel insights into the response of Atlantic salmon (Salmo salar) to Piscirickettsia salmonis: Interplay of coding genes and lncRNAs during bacterial infection. Fish Shellfish Immunol. 2016, 59, 427–438. [Google Scholar] [CrossRef]
- Michels, K.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin and Host Defense against Infectious Diseases. PLoS Pathog. 2015, 11, e1004998. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.K.; Jeong, J.H.; Lee, J.M.; Kim, K.S.; Park, S.H.; Kim, Y.D.; Koh, M.; Shin, M.; Jung, Y.S.; Kim, H.S.; et al. Inverse agonist of estrogen-related receptor γ controls Salmonella Typhimurium infection by modulating host iron homeostasis. Nat. Med. 2014, 20, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Cortés, M.; Sánchez, P.; Ruiz, P.; Haro, R.; Sáez, J.; Sánchez, F.; Hernández, M.; Oliver, C.; Yáñez, A.J. In vitro expression of Sec-dependent pathway and type 4B secretion system in Piscirickettsia salmonis. Microb. Pathog. 2017, 110, 586–593. [Google Scholar] [CrossRef]
- Gómez, F.A.; Tobar, J.A.; Henríquez, V.; Sola, M.; Altamirano, C.; Marshall, S.H. Evidence of the presence of a functional Dot/Icm type IV-B secretion system in the fish bacterial pathogen Piscirickettsia salmonis. PLoS ONE 2013, 8, e54934. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SHK-1 Cells | ASK Cells | Zebrafish Kidney Cells | |
|---|---|---|---|
| Actin P. salmonis DNA |  |  |  |
| Cell type |
|
|
|
| Cellular viability after P. salmonis infection |
|
|
|
| Phenotypic effects of P. salmonis infection in cell cultures |
|
|
|
| Immune response to P. salmonis infection |
|
* IL-8 and IL-10 did not change their expression. |
* IL-8 did not change its expression. |
| P. salmonis intracellular replication |
|
|
|
| P. salmonis genes induced during infection |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Severín, J.; Tandberg, J.I.; Winther-Larsen, H.C.; Chávez, F.P.; Cambiazo, V. Comparative Analysis of Salmon Cell Lines and Zebrafish Primary Cell Cultures Infection with the Fish Pathogen Piscirickettsia salmonis. Microorganisms 2021, 9, 2516. https://doi.org/10.3390/microorganisms9122516
Ortiz-Severín J, Tandberg JI, Winther-Larsen HC, Chávez FP, Cambiazo V. Comparative Analysis of Salmon Cell Lines and Zebrafish Primary Cell Cultures Infection with the Fish Pathogen Piscirickettsia salmonis. Microorganisms. 2021; 9(12):2516. https://doi.org/10.3390/microorganisms9122516
Chicago/Turabian StyleOrtiz-Severín, Javiera, Julia I. Tandberg, Hanne C. Winther-Larsen, Francisco P. Chávez, and Verónica Cambiazo. 2021. "Comparative Analysis of Salmon Cell Lines and Zebrafish Primary Cell Cultures Infection with the Fish Pathogen Piscirickettsia salmonis" Microorganisms 9, no. 12: 2516. https://doi.org/10.3390/microorganisms9122516
APA StyleOrtiz-Severín, J., Tandberg, J. I., Winther-Larsen, H. C., Chávez, F. P., & Cambiazo, V. (2021). Comparative Analysis of Salmon Cell Lines and Zebrafish Primary Cell Cultures Infection with the Fish Pathogen Piscirickettsia salmonis. Microorganisms, 9(12), 2516. https://doi.org/10.3390/microorganisms9122516