
Insights into the Neutralization and DNA Binding of Toxin–Antitoxin System ParESO-CopASO by Structure-Function Studies

Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of Plasmids and Bacterial Strains
2.2. Protein Expression and Purification
2.3. Protein Crystallization and Structure Determination
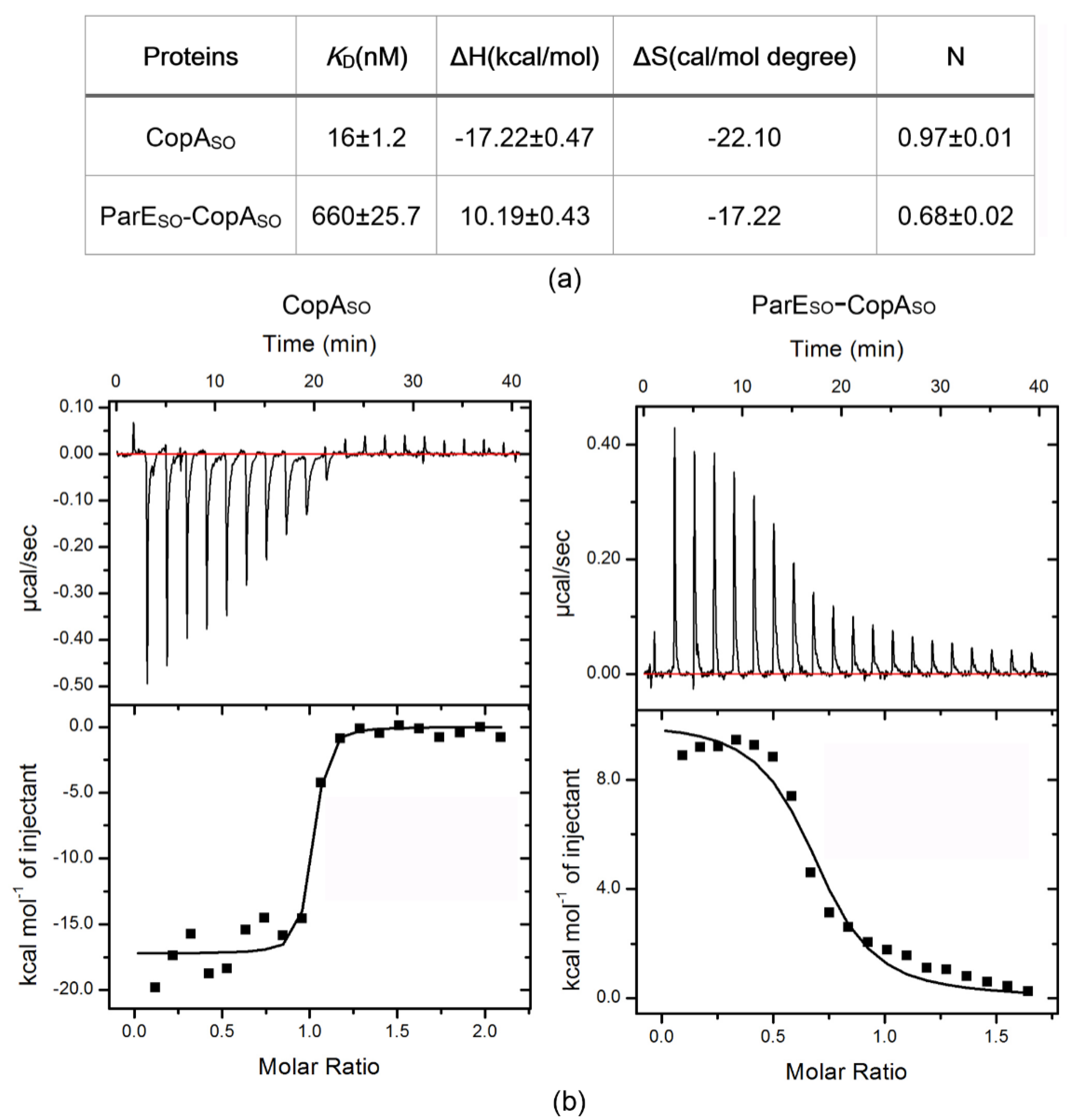
2.4. Isothermal Titration Calorimetry (ITC)
2.5. Cell Toxicity Assay
3. Results
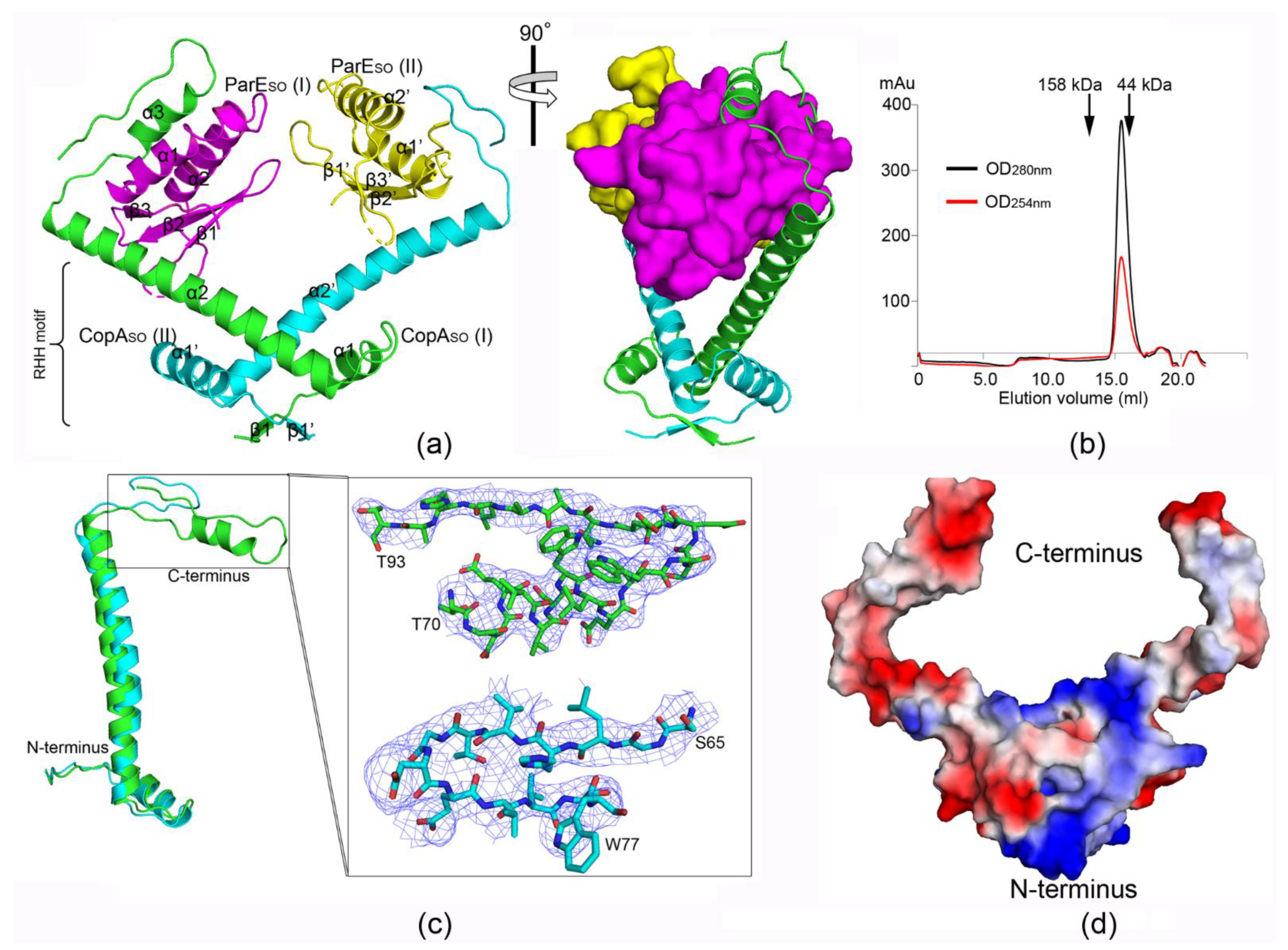
3.1. ParESO-CopASO Complex Has an Open V-shaped Structure
3.2. CopASO Adopts a Partially Folded C-Terminal Conformation upon ParESO Binding
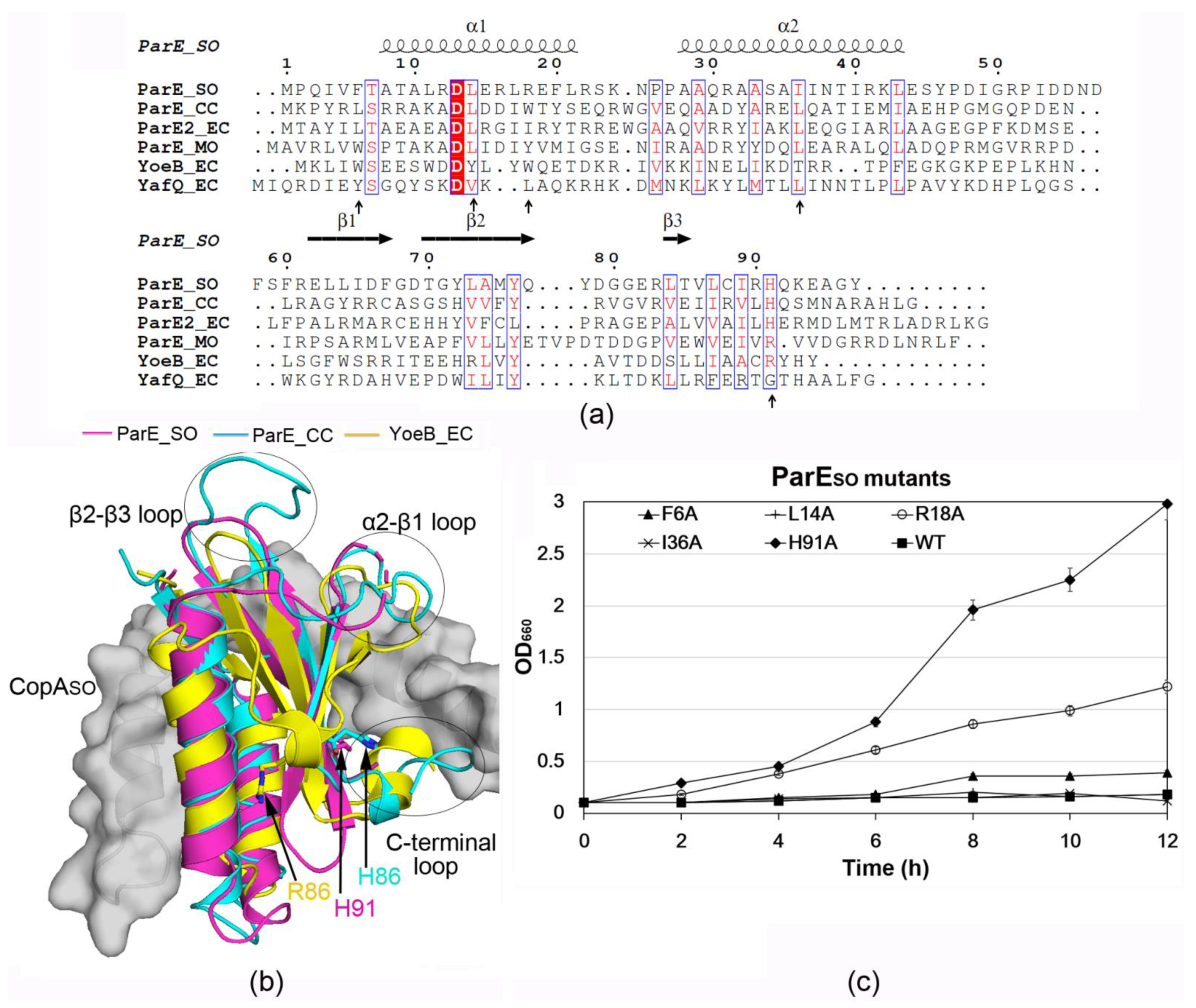
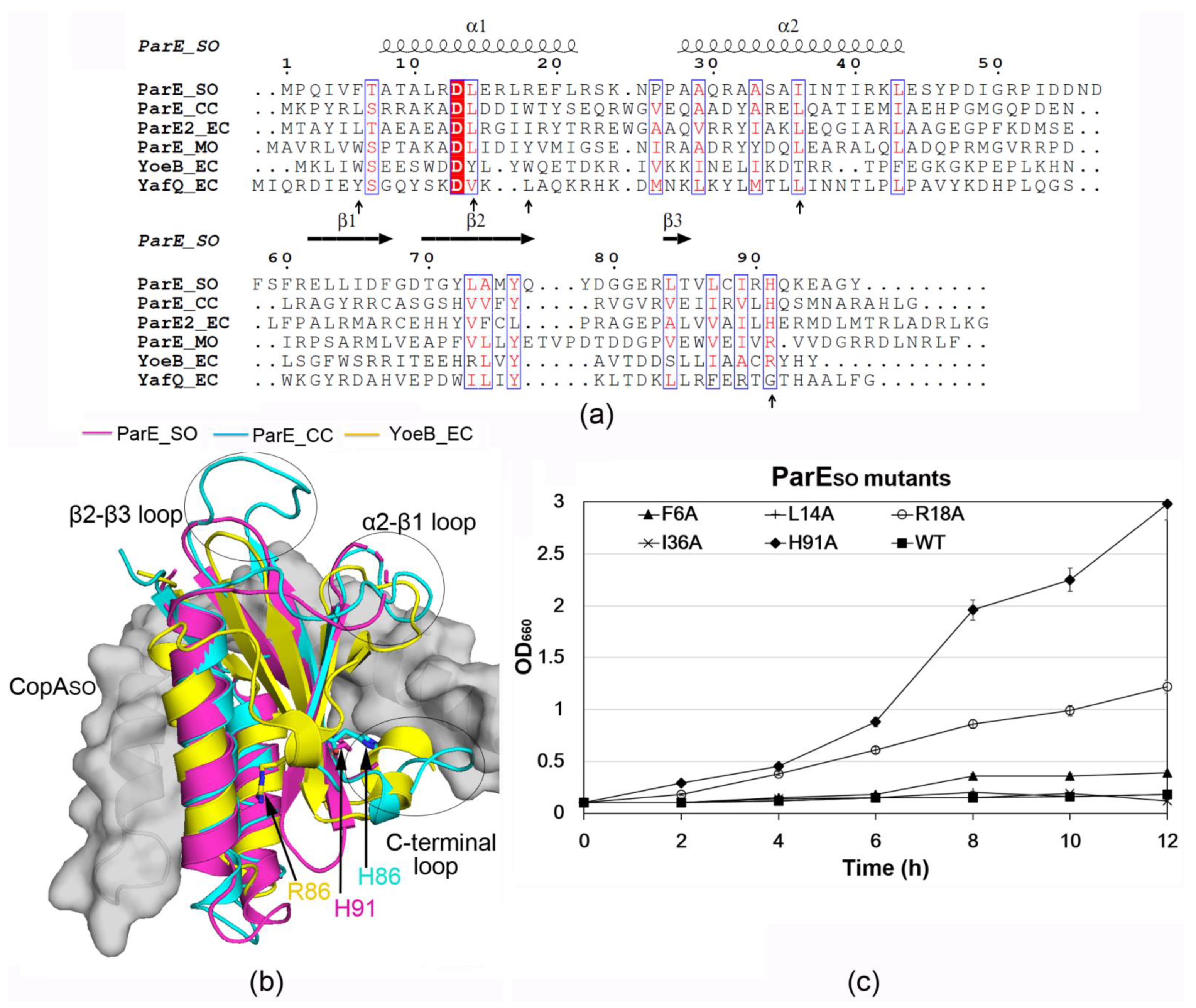
3.3. ParESO Is Structurally Homologous to ParE/RelE Toxins
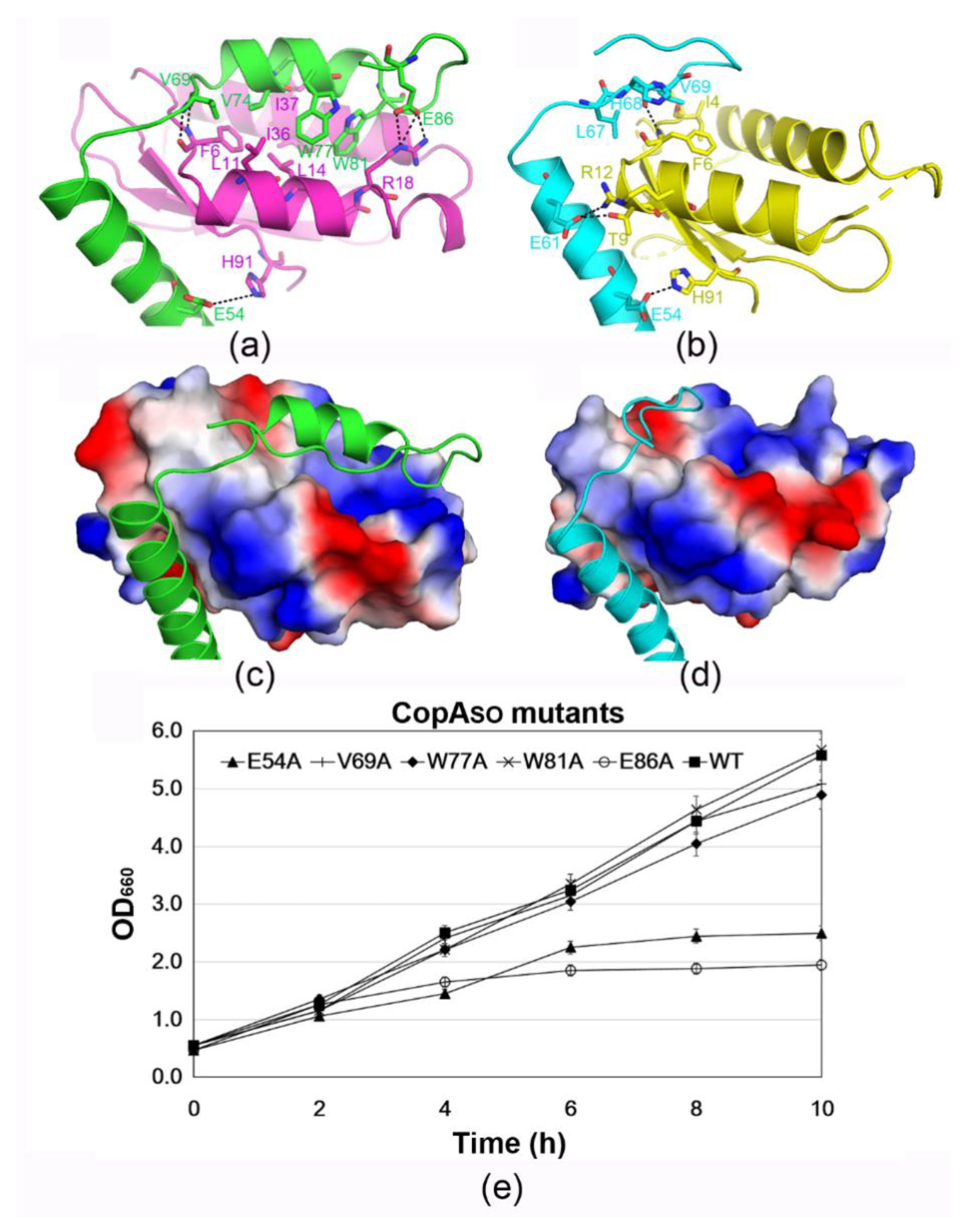
3.4. The C-Terminal Extension of CopASO Binds into a Positive and Hydrophobic Groove of ParESO
3.5. H91 Is a Key Residue for ParESO Toxicity
3.6. DNA-Binding Capacity of CopASO Is Inhibited upon ParESO Binding
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gerdes, K.; Christensen, S.K.; Løbner-Olesen, A. Prokaryotic toxin–antitoxin stress response loci. Nat. Rev. Genet. 2005, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Inouye, M. Regulation of growth and death in Escherichia coli by toxin–antitoxin systems. Nat. Rev. Genet. 2011, 9, 779–790. [Google Scholar] [CrossRef]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef]
- Wang, X.; Yao, J.; Sun, Y.C.; Wood, T.K. Type VII toxin/antitoxin classification system for antitoxins that enzymatically neutralize toxins. Trends Microbiol. 2021, 29, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Gao, X.; Zhu, K.; Yin, H.; Mao, X.; Wojdyla, J.A.; Qin, B.; Huang, H.; Wang, M.; Sun, Y.C.; et al. Characterization of a toxin-antitoxin system in Mycobacterium tuberculosis suggests neutralization by phosphorylation as the antitoxicity mech-anism. Commun. Biol. 2020, 3, 216. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhen, X.; Tang, K.; Liu, T.; Xu, X.; Chen, Z.; Guo, Y.; Liu, X.; Wood, T.K.; Ouyang, S.; et al. Novel polyad-enylylationdependent neutralization mechanism of the HEPN/MNT toxin/antitoxin system. Nucleic Acids Res. 2020, 48, 11054–11067. [Google Scholar] [CrossRef]
- Loris, R.; Garcia-Pino, A. Disorder- and Dynamics-Based Regulatory Mechanisms in Toxin–Antitoxin Modules. Chem. Rev. 2014, 114, 6933–6947. [Google Scholar] [CrossRef]
- Anantharaman, V.; Aravind, L. New connections in the prokaryotic toxin-antitoxin network: Relationship with the eukaryotic nonsense-mediated RNA decay system. Genome Biol. 2003, 4, R81. [Google Scholar] [CrossRef] [Green Version]
- Dalton, K.M.; Crosson, S. A conserved mode of protein recognition and binding in a ParD-ParE toxin-antitoxin complex. Biochemistry 2010, 49, 2205–2215. [Google Scholar] [CrossRef] [Green Version]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef]
- Gupta, M.; Nayyar, N.; Chawla, M.; Sitaraman, R.; Bhatnagar, R.; Banerjee, N. The Chromosomal parDE2 Tox-in-Antitoxin System of Mycobacterium tuberculosis H37Rv: Genetic and Functional Characterization. Front. Microbiol. 2016, 7, 886. [Google Scholar] [CrossRef] [Green Version]
- Sterckx, Y.G.J.; Jové, T.; Shkumatov, A.V.; Garcia-Pino, A.; Geerts, L.; De Kerpel, M.; Lah, J.; De Greve, H.; Van Melderen, L.; Loris, R. A unique hetero-hexadecameric architecture displayed by the Escherichia coli O157 PaaA2-ParE2 antitoxin-toxin complex. J. Mol. Biol. 2016, 428, 1589–1603. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Pogliano, J.; Helinski, D.R.; Konieczny, I. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 2002, 44, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.K.; Gerdes, K. RelE toxins from Bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 2003, 48, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.M.; Woychik, N.A. Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. J. Biol. Chem. 2009, 284, 18605–18613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Inouye, M. The inhibitory mechanism of protein synthesis by YoeB, an Escherichia coli toxin. J. Bacteriol. 2009, 284, 6627–6638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, C.; Gao, Y.-G.; Andersen, K.R.; Dunham, C.M.; Kelley, A.C.; Hentschel, J.; Gerdes, K.; Ramakrishnan, V.; Brodersen, D.E. The Structural Basis for mRNA Recognition and Cleavage by the Ribosome-Dependent Endonuclease RelE. Cell 2009, 139, 1084–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, K.; Zavialov, A.; Pavlov, M.Y.; Elf, J.; Gerdes, K.; Ehrenberg, M. The Bacterial Toxin RelE Displays Codon-Specific Cleavage of mRNAs in the Ribosomal A Site. Cell 2003, 112, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Guo, Y.; Wang, P.; Zeng, Z.; Li, B.; Tang, K.; Liu, X.; Wang, X. Type II toxin/antitoxin system ParESO/CopASO stabilizes prophage CP4So in Shewanella oneidensis. Environ. Microbiol. 2018, 20, 1224–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Li, Q.; Zhang, J.; Li, F.; Yao, J.; Zhang, J.; Liu, L.; Wang, X.; Zhang, X. Structure and allosteric coupling of type II antitoxin CopASO. Biochem. Biophs. Res. Commun. 2019, 514, 1122–1127. [Google Scholar] [CrossRef]
- Wan, H.; Otsuka, Y.; Gao, Z.; Wei, Y.; Chen, Z.; Masuda, M.; Yonesaki, T.; Zhang, H.; Dong, Y. Structural insights into the inhibition mechanism of bacterial toxin LsoA by bacteriophage antitoxin Dmd. Mol. Microbiol. 2016, 101, 757–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C. Maximum-likelihood density modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1002–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunko’ czi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.; Cowtan, K.D. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aakre, C.D.; Herrou, J.; Phung, T.N.; Perchuk, B.S.; Crosson, S.; Laub, M.T. Evolving new protein-protein in-teraction specificity through promiscuous intermediates. Cell 2015, 163, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Kamada, K.; Hanaoka, F. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin Mol. Cell 2005, 19, 497–509. [Google Scholar]
- Liang, Y.; Gao, Z.; Wang, F.; Zhang, Y.; Dong, Y.; Liu, Q. Structural and functional characterization of Escherichia coli toxin-antitoxin complex DinJ-YafQ. J. Biol. Chem. 2014, 289, 21191–21202. [Google Scholar] [CrossRef] [Green Version]
- Francuski, D.; Saenger, W. Crystal structure of the antitoxin-toxin protein complex RelB-RelE from Methanococcus jannaschii. J. Mol. Biol. 2014, 393, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Muthuramalingam, M.; White, J.C.; Murphy, T.; Ames, J.R.; Bourne, C.R. The toxin from a ParDE toxin-antitoxin system found in Pseudomonas aeruginosa offers protection to cells challenged with anti-gyrase antibiotics. Mol. Microbiol. 2018, 111, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Rodriguez, G.; Girardin, Y.; Volkov, A.N.; Singh, R.K.; Muruganandam, G.; Van Dyck, J.; Sobott, F.; Versées, W.; Charlier, D.; Loris, R. Entropic pressure controls the oligomerization of the Vibrio cholerae ParD2 antitoxin. Acta Crystallogr. Sect. D Struct. Biol. 2021, 77, 904–920. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, M.; Borch, J.; Jørgensen, M.G.; Gerdes, K. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 2008, 69, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Cataudella, I.; Trusina, A.; Sneppen, K.; Gerdes, K.; Mitarai, N. Conditional cooperativity in toxin-antitoxin reg-ulation prevents random toxin activation and promotes fast translational recovery. Nucleic Acids Res. 2012, 40, 6424–6434. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Kang, S.-M.; Park, S.J.; Jin, C.; Yoon, H.-J.; Lee, B.-J. Functional insights into the Streptococcus pneumoniae HicBA toxin–antitoxin system based on a structural study. Nucleic Acids Res. 2018, 46, 6371–6386. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | ParESO-CopASO Complex (Se-Met) |
|---|---|
| Beamline | SSRF 17U1 |
| Wavelength (Å) | 0.9788 |
| Space group | P43212 |
| Unit-cell parameters | a = 85.4 Å, b = 85.4 Å, c = 110.9 Å, α = β = γ = 90° |
| Resolution (Å) | 3.80 (3.87–3.80) a |
| Number of unique reflections | 7720 (375) |
| Completeness (%) | 100 (99.7) |
| Redundancy | 20.3 (22.8) |
| Mean I/ơ (I) | 25.0 (7.2) |
| Molecules in asymmetric unit | 4 |
| Rmerge (%) | 19.4 (69.6) |
| Rmeas (%) | 19.7 (71.2) |
| CC1/2 | 100 (98.2) |
| Structure Refinement | |
| Reflections used in refinement | 7667 |
| Resolution range (Å) | 33.93–3.80 |
| Rwork/Rfree (%) | 29.8/30.5 |
| Protein atoms | 2618 |
| Protein residues | 312 |
| Average B factor (Å2) | 133.8 |
| Ramachandran plot (%) | |
| Most favored | 90.1 |
| Allowed | 6.6 |
| Disallowed | 3.6 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.806 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Du, X.-J.; Liu, Y.; Gao, Z.-Q.; Geng, Z.; Dong, Y.-H.; Zhang, H. Insights into the Neutralization and DNA Binding of Toxin–Antitoxin System ParESO-CopASO by Structure-Function Studies. Microorganisms 2021, 9, 2506. https://doi.org/10.3390/microorganisms9122506
Zhou J, Du X-J, Liu Y, Gao Z-Q, Geng Z, Dong Y-H, Zhang H. Insights into the Neutralization and DNA Binding of Toxin–Antitoxin System ParESO-CopASO by Structure-Function Studies. Microorganisms. 2021; 9(12):2506. https://doi.org/10.3390/microorganisms9122506
Chicago/Turabian StyleZhou, Juan, Xue-Jian Du, Ying Liu, Zeng-Qiang Gao, Zhi Geng, Yu-Hui Dong, and Heng Zhang. 2021. "Insights into the Neutralization and DNA Binding of Toxin–Antitoxin System ParESO-CopASO by Structure-Function Studies" Microorganisms 9, no. 12: 2506. https://doi.org/10.3390/microorganisms9122506
APA StyleZhou, J., Du, X.-J., Liu, Y., Gao, Z.-Q., Geng, Z., Dong, Y.-H., & Zhang, H. (2021). Insights into the Neutralization and DNA Binding of Toxin–Antitoxin System ParESO-CopASO by Structure-Function Studies. Microorganisms, 9(12), 2506. https://doi.org/10.3390/microorganisms9122506