
Assembly of a Benthic Microbial Community in a Eutrophic Bay with a Long History of Oyster Culturing



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Sites and Sample Collection
2.2. Environmental Parameters
2.3. DNA Extraction, PCR Amplification, Pyrosequencing, and Data Processing
2.4. Data Analysis
2.4.1. Environmental Parameters
2.4.2. Community Biodiversity
2.4.3. Deterministic and Stochastic Processes
2.4.4. Network Analysis
2.4.5. RDA and Model Selection
2.4.6. Water Parameters and Sedimental Acid Volatile Sulfides (AVS)
2.4.7. Genome Prediction
3. Results
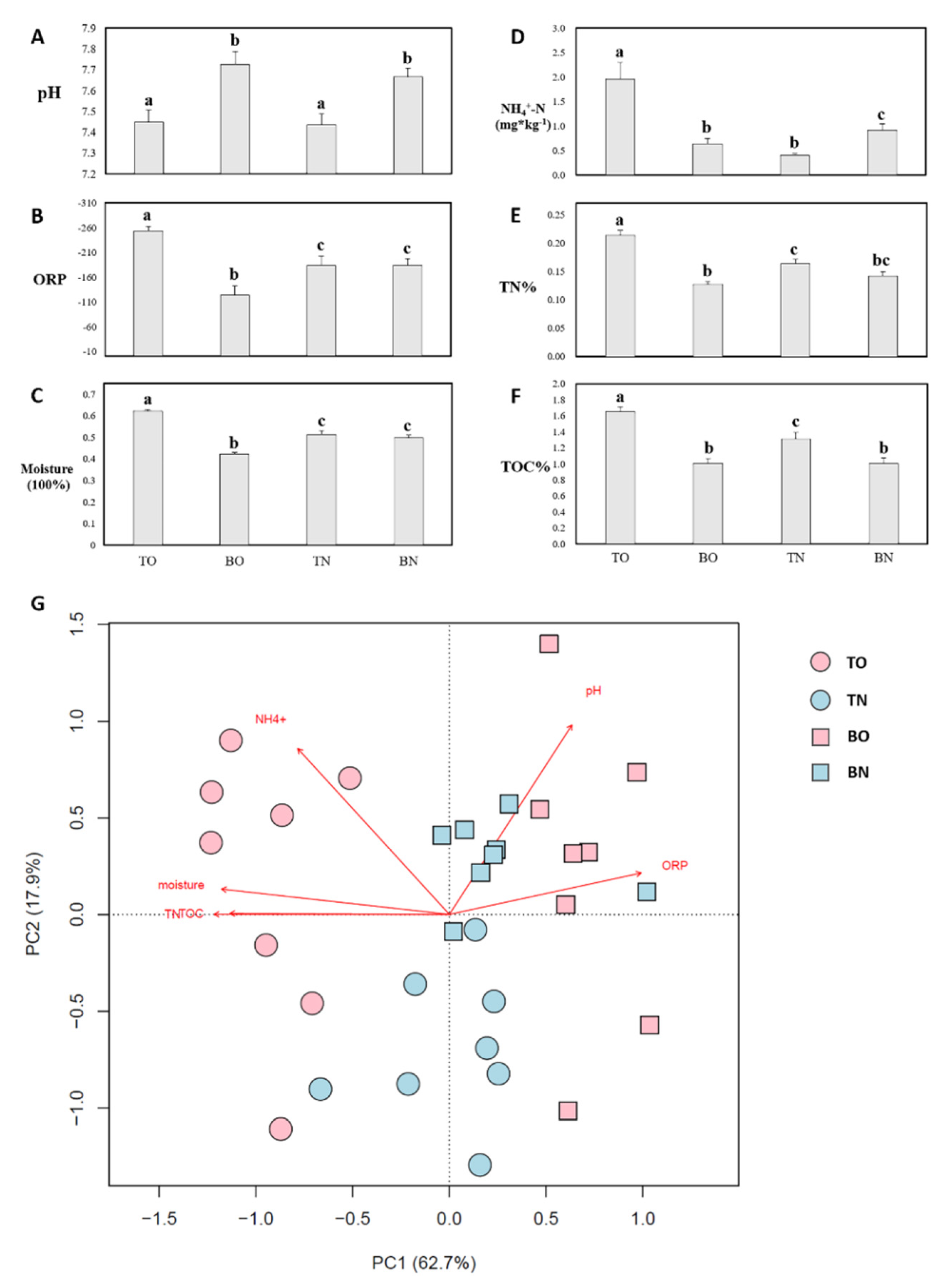
3.1. Environmental Parameters
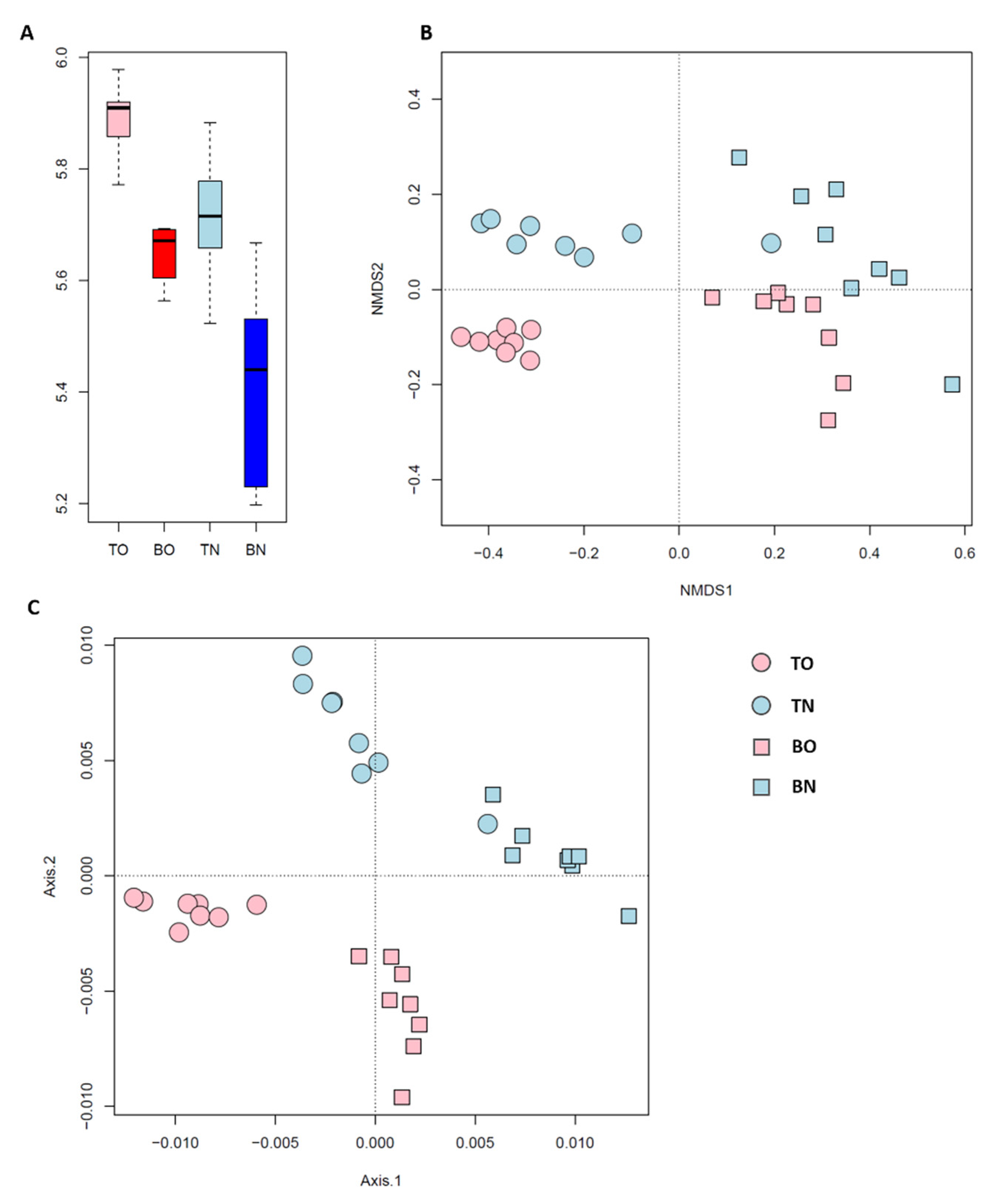
3.2. Community Composition and Biodiversity
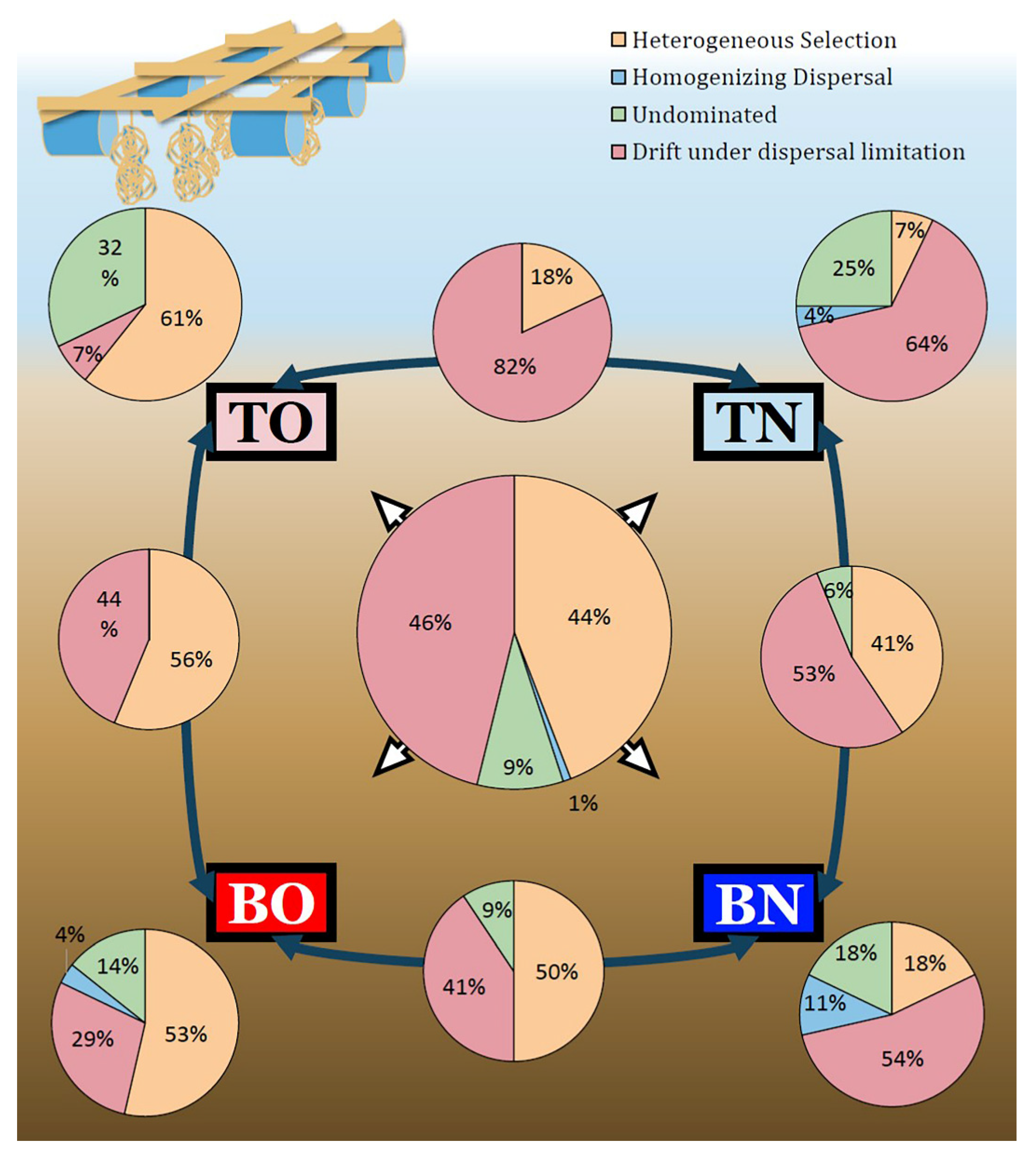
3.3. Deterministic vs. Stochastic Processes
3.4. Network and Keystone Species
3.5. Keystone Species vs. Other Species
3.6. Model Selection
3.7. Water Parameters and Sedimental AVS
3.8. Genome Predicion of TO Community and Keystone Species Based on KEGG Database
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryther, J.H.; Dunstan, W.M. Nitrogen, phosphorus, and eutrophication in the coastal marine environment. Science 1971, 171, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R. Eutrophication—The Future Marine Coastal Nuisance. Mar. Pollut. Bull. 1985, 16, 227–231. [Google Scholar] [CrossRef]
- Smith, V.; Tilman, G.; Nekola, J. Eutrophication: Impacts of excess nutrient inputs on freshwater, marine, and terrestrial ecosystems. Environ. Pollut. 1999, 100, 179–196. [Google Scholar] [CrossRef]
- Smith, V.H.; Schindler, D.W. Eutrophication science: Where do we go from here? Trends Ecol. Evol. 2009, 24, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Hoellein, T.J.; Zarnoch, C.B. Effect of eastern oysters (Crassostrea virginica) on sediment carbon and nitrogen dynamics in an urban estuary. Ecol. Appl. 2014, 24, 271–286. [Google Scholar] [CrossRef]
- Chakraborty, P. Oyster reef restoration in controlling coastal pollution around India: A viewpoint. Mar. Pollut. Bull. 2017, 115, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.E.; Emery, K.A.; Anderson, I.C.; Pace, M.L.; Brush, M.J.; Rheuban, J.E. Quantifying the Effects of Commercial Clam Aquaculture on C and N Cycling: An Integrated Ecosystem Approach. Chesap. Sci. 2016, 39, 1746–1761. [Google Scholar] [CrossRef]
- Nizzoli, D.; Welsh, D.T.; Fano, E.A.; Viaroli, P. Impact of clam and mussel farming on benthic metabolism and nitrogen cycling, with emphasis on nitrate reduction pathways. Mar. Ecol. Prog. Ser. 2006, 315, 151–165. [Google Scholar] [CrossRef]
- Hoellein, T.J.; Zarnoch, C.B.; Bruesewitz, D.A.; DeMartini, J. Contributions of freshwater mussels (Unionidae) to nutrient cycling in an urban river: Filtration, recycling, storage, and removal. Biogeochemistry 2017, 135, 307–324. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, S.; Liu, Y.; Yang, H. Biologically Induced Deposition of Fine Suspended Particles by Filter-Feeding Bivalves in Land-Based Industrial Marine Aquaculture Wastewater. PLoS ONE 2014, 9, e107798. [Google Scholar] [CrossRef]
- Sebastiano, D.; Levinton, J.S.; Doall, M.; Kamath, S. Using a Shellfish Harvest Strategy to Extract High Nitrogen Inputs in Urban and Suburban Coastal Bays: Practical and Economic Implications. J. Shellfish. Res. 2015, 34, 573–583. [Google Scholar] [CrossRef]
- Welsh, D.; Nizzoli, D.; Fano, E.A.; Viaroli, P. Direct contribution of clams (Ruditapes philippinarum) to benthic fluxes, nitrification, denitrification and nitrous oxide emission in a farmed sediment. Estuar. Coast. Shelf Sci. 2015, 154, 84–93. [Google Scholar] [CrossRef]
- Caffrey, J.M.; Hollibaugh, J.T.; Mortazavi, B. Living oysters and their shells as sites of nitrification and denitrification. Mar. Pollut. Bull. 2016, 112, 86–90. [Google Scholar] [CrossRef]
- Smyth, A.R.; Piehler, M.F.; Grabowski, J.H. Habitat context influences nitrogen removal by restored oyster reefs. J. Appl. Ecol. 2015, 52, 716–725. [Google Scholar] [CrossRef]
- Smyth, A.R.; Murphy, A.E.; Anderson, I.C.; Song, B. Differential Effects of Bivalves on Sediment Nitrogen Cycling in a Shallow Coastal Bay. Chesap. Sci. 2017, 41, 1147–1163. [Google Scholar] [CrossRef]
- Murphy, A.E.; Kolkmeyer, R.; Song, B.; Anderson, I.C.; Bowen, J. Bioreactivity and Microbiome of Biodeposits from Filter-Feeding Bivalves. Microb. Ecol. 2019, 77, 343–357. [Google Scholar] [CrossRef]
- Feinman, S.G.; Farah, Y.R.; Bauer, J.M.; Bowen, J.L. The Influence of Oyster Farming on Sediment Bacterial Communities. Chesap. Sci. 2017, 41, 800–814. [Google Scholar] [CrossRef]
- Salazar, G.; Sunagawa, S. Marine microbial diversity. Curr. Biol. 2017, 27, R489–R494. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Xu, S.; Qi, Z.; Zhu, Q.; Huang, H.; Weber, F. Influence of suspended mariculture on vertical distribution profiles of bacteria in sediment from Daya Bay, Southern China. Mar. Pollut. Bull. 2019, 146, 816–826. [Google Scholar] [CrossRef]
- Woodcock, S.; Van Der Gast, C.J.; Bell, T.; Lunn, M.; Curtis, T.P.; Head, I.M.; Sloan, W.T. Neutral assembly of bacterial communities. Fems Microbiol. Ecol. 2007, 62, 171–180. [Google Scholar] [CrossRef]
- Vellend, M. Conceptual Synthesis in Community Ecology. Q. Rev. Biol. 2010, 85, 183–206. [Google Scholar] [CrossRef]
- Chase, J.M.; Kraft, N.; Smith, K.G.; Vellend, M.; Inouye, B.D. Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere 2011, 2, art24-11. [Google Scholar] [CrossRef]
- Stegen, J.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef]
- Yao, Z.; Du, S.; Liang, C.; Zhao, Y.; Dini-Andreote, F.; Wang, K.; Zhang, D. Bacterial Community Assembly in a Typical Estuarine Marsh with Multiple Environmental Gradients. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef] [PubMed]
- Plante, C.J.; Fleer, V.; Jones, M.L. Neutral processes and species sorting in benthic microalgal community assembly: Effects of tidal resuspension. J. Phycol. 2016, 52, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Li, A.Z.; Han, X.B.; Zhang, M.X.; Zhou, Y.; Chen, M.; Yao, Q.; Zhu, H.H. Culture-Dependent and -Independent Analyses Reveal the Diversity, Structure, and Assembly Mechanism of Benthic Bacterial Community in the Ross Sea, Antarctica. Front. Microbiol. 2019, 10, 2523. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; Van Der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Genet. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Yan, H.; He, X.; Lei, Y.; Wang, Y.; Su, H.; Jiang, S. Land use-induced change in trophic state of Shenzhen Bay (South China) over the past half-century. Mar. Pollut. Bull. 2019, 145, 208–213. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, L.; Deng, Y.; Zhi, X.; Jiang, Y.-H.; Tu, Q.; Xie, J.; Van Nostrand, J.; He, Z.; Yang, Y. Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 2011, 5, 1303–1313. [Google Scholar] [CrossRef]
- Ren, Y.; Niu, J.; Huang, W.; Peng, D.; Xiao, Y.; Zhang, X.; Liang, Y.; Liu, X.; Yin, H. Comparison of microbial taxonomic and functional shift pattern along contamination gradient. BMC Microbiol. 2016, 16, 110. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Zeng, S.; Liu, J.; Yan, M.; Weng, S.; He, J.; Huang, Z. Characterization of Prokaryotic and Eukaryotic Microbial Community in PacificWhite Shrimp Ponds. J. Aquacult. Res. Dev. 2016, 7, 463–472. [Google Scholar] [CrossRef]
- Hung, C.-C.; Tseng, C.-W.; Gong, G.-C.; Chen, K.-S.; Chen, M.-H.; Hsu, S.-C. Fluxes of particulate organic carbon in the East China Sea in summer. Biogeosciences 2013, 10, 6469–6484. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Tripathi, B.M.; Stegen, J.C.; Kim, M.; Dong, K.; Adams, J.M.; Lee, Y.K. Soil pH mediates the balance between stochastic and deterministic assembly of bacteria. ISME J. 2018, 12, 1072–1083. [Google Scholar] [CrossRef]
- Stegen, J.; Lin, X.; Fredrickson, J.K.; Chen, X.; Kennedy, D.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Meinicke Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, M.L.; Smyth, A.R.; Luckenbach, M.W.; Carmichael, R.H.; Brown, B.L.; Cornwell, J.C.; Piehler, M.F.; Owens, M.S.; Dalrymple, D.J.; Higgins, C.B. Use of oysters to mitigate eutrophication in coastal waters. Estuar. Coast. Shelf Sci. 2014, 151, 156–168. [Google Scholar] [CrossRef]
- Smith, V.H. Eutrophication of freshwater and coastal marine ecosystems—A global problem. Environ. Sci. Pollut. Res. 2003, 10, 126–139. [Google Scholar] [CrossRef]
- Zhang, S.; Mao, X.Z. Hydrology, sediment circulation and long-term morphological changes in highly urbanized Shenzhen River estuary, China: A combined field experimental and modeling approach. J. Hydrol. 2015, 529, 1562–1577. [Google Scholar] [CrossRef]
- Leon, S.M.S.S.-D.; Okunishi, S.; Kihira, M.; Nakano, M.; Nunal, S.N.; Hidaka, M.; Yoshikawa, T.; Maeda, H. Characterization of the Bacterial Community in the Sediment of a Brackish Lake with Oyster Aquaculture. Biocontrol Sci. 2013, 18, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.T.E.; Fulweiler, R.W.; Chowdhury, P.R. 16S rRNA Amplicon Sequencing of Sediment Bacterial Communities in an Oyster Farm in Rhode Island. Microbiol. Resour. Announc. 2019, 8, e01074-19. [Google Scholar] [CrossRef] [PubMed]
- Stegen, J.; Fredrickson, J.K.; Wilkins, M.J.; Konopka, A.E.; Nelson, W.; Arntzen, E.V.; Chrisler, W.B.; Chu, R.K.; Danczak, R.E.; Fansler, S.J.; et al. Groundwater–surface water mixing shifts ecological assembly processes and stimulates organic carbon turnover. Nat. Commun. 2016, 7, 11237. [Google Scholar] [CrossRef]
- Chase, J.M.; Myers, J.A. Disentangling the importance of ecological niches from stochastic processes across scales. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, Q.; Tian, T.; Li, D.; Cheng, G.; Mu, J.; Wu, Q.; Niu, F.; Stegen, J.; An, L.; et al. Relative Roles of Deterministic and Stochastic Processes in Driving the Vertical Distribution of Bacterial Communities in a Permafrost Core from the Qinghai-Tibet Plateau, China. PLoS ONE 2015, 10, e0145747. [Google Scholar] [CrossRef] [PubMed]
- Valverde, A.; Makhalanyane, T.; Cowan, D.A. Contrasting assembly processes in a bacterial metacommunity along a desiccation gradient. Front. Microbiol. 2014, 5, 668. [Google Scholar] [CrossRef]
- Geng, H.; Tran-Gyamfi, M.B.; Lane, T.W.; Sale, K.L.; Yu, E. Changes in the Structure of the Microbial Community Associated with Nannochloropsis salina following Treatments with Antibiotics and Bioactive Compounds. Front. Microbiol. 2016, 7, 1155. [Google Scholar] [CrossRef]
- Liao, H.; Yen, J.Y.; Guan, Y.; Ke, D.; Liu, C. Differential responses of stream water and bed sediment microbial communities to watershed degradation. Environ. Int. 2019, 134, 105198. [Google Scholar] [CrossRef]
- Ji, Y.; Angel, R.; Klose, M.; Claus, P.; Marotta, H.; Pinho, L.; Enrich-Prast, A.; Conrad, R. Structure and function of methanogenic microbial communities in sediments of Amazonian lakes with different water types. Environ. Microbiol. 2016, 18, 5082–5100. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Zhang, Y.; Ning, D.; Su, Z.; Tang, Y.; Huang, B.; Mu, Q.; Wen, D. Dynamics of Sediment Microbial Functional Capacity and Community Interaction Networks in an Urbanized Coastal Estuary. Front. Microbiol. 2018, 9, 2731. [Google Scholar] [CrossRef] [PubMed]
- Matturro, B.; Viggi, C.C.; Aulenta, F.; Rossetti, S. Cable Bacteria and the Bioelectrochemical Snorkel: The Natural and Engineered Facets Playing a Role in Hydrocarbons Degradation in Marine Sediments. Front. Microbiol. 2017, 8, 952. [Google Scholar] [CrossRef]
- Mußmann, M.; Pjevac, P.; Krüger, K.; Dyksma, S. Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J. 2017, 11, 1276–1281. [Google Scholar] [CrossRef]
- Yan, Z.; Hao, Z.; Wu, H.; Jiang, H.; Yang, M.; Wang, C. Co-occurrence patterns of the microbial community in polycyclic aromatic hydrocarbon-contaminated riverine sediments. J. Hazard. Mater. 2018, 367, 99–108. [Google Scholar] [CrossRef]
- Han, X.; Wang, F.; Zhang, D.; Feng, T.; Zhang, L. Nitrate-assisted biodegradation of polycyclic aromatic hydrocarbons (PAHs) in the water-level-fluctuation zone of the three Gorges Reservoir, China: Insights from in situ microbial interaction analyses and a microcosmic experiment. Environ. Pollut. 2020, 268, 115693. [Google Scholar] [CrossRef]
- Qiu, Y.-W.; Zhang, G.; Liu, G.-Q.; Guo, L.-L.; Li, X.-D.; Wai, O. Polycyclic aromatic hydrocarbons (PAHs) in the water column and sediment core of Deep Bay, South China. Estuar. Coast. Shelf Sci. 2009, 83, 60–66. [Google Scholar] [CrossRef]
- Muwawa, E.M.; Obieze, C.C.; Makonde, H.M.; Jefwa, J.M.; Kahindi, J.H.P.; Khasa, D.P. 16S rRNA gene amplicon-based metagenomic analysis of bacterial communities in the rhizospheres of selected mangrove species from Mida Creek and Gazi Bay, Kenya. PLoS ONE 2021, 16, e0248485. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| βNTI | RCbray | |||||
|---|---|---|---|---|---|---|
| PC2 | PC24 | PC8 | PC9 | PC5 | PC11 | |
| Environmental parameters | ||||||
| Moisture | −0.1060 | −0.0086 | 0.0457 | −0.0512 | −0.7764 | −0.0177 |
| NH4+ | −0.6505 | 0.0077 | −0.0378 | −0.0210 | 0.3524 | 0.0147 |
| ORP | −0.1505 | −0.0074 | −0.0674 | −0.0279 | −0.3272 | −0.0171 |
| pH | −0.7301 | 0.0036 | −0.0538 | 0.0329 | −0.1329 | 0.0037 |
| TN | −0.0044 | 0.0097 | −0.2756 | −0.0019 | −0.0968 | −0.0316 |
| TOC | −0.0033 | −0.0055 | 0.3042 | −0.0048 | 0.3193 | 0.0386 |
| Keystone species | ||||||
| OTU1 | −0.0339 | −0.1206 | 0.5251 | 0.3698 | −0.1035 | −0.0057 |
| Syntrophobacteraceae | ||||||
| OTU1764 | 0.0034 | 0.2667 | −0.0029 | −0.0150 | −0.0196 | 0.0396 |
| Pirellulaceae | ||||||
| OTU18 | 0.0327 | 0.1811 | −0.1745 | 0.1694 | −0.0149 | −0.2999 |
| B2M28 * | ||||||
| OTU1866 | −0.0083 | 0.2450 | 0.0360 | 0.0331 | −0.0024 | −0.0653 |
| ADurb.Bin180 * | ||||||
| OTU209 | 0.0139 | −0.0688 | −0.1956 | 0.2805 | 0.0142 | 0.1885 |
| Bacteroidetes_BD2-2 | ||||||
| OTU4 | 0.0177 | −0.0827 | −0.0212 | −0.5950 | −0.0233 | −0.0096 |
| Anaerolineaceae | ||||||
| OTU44 | 0.0207 | −0.0469 | −0.2507 | 0.0377 | 0.0344 | −0.4426 |
| Sulfurovaceae | ||||||
| OTU47 | 0.0132 | −0.3834 | −0.1244 | 0.1284 | 0.0185 | −0.2602 |
| Chromatiaceae | ||||||
| OTU890 | −0.0058 | −0.0061 | −0.0207 | 0.0891 | 0.0257 | −0.2895 |
| SBR1031 * | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.; Song, J.; Yan, Q.; Zhou, J.; Cai, Z. Assembly of a Benthic Microbial Community in a Eutrophic Bay with a Long History of Oyster Culturing. Microorganisms 2021, 9, 2019. https://doi.org/10.3390/microorganisms9102019
Song X, Song J, Yan Q, Zhou J, Cai Z. Assembly of a Benthic Microbial Community in a Eutrophic Bay with a Long History of Oyster Culturing. Microorganisms. 2021; 9(10):2019. https://doi.org/10.3390/microorganisms9102019
Chicago/Turabian StyleSong, Xiao, Junting Song, Qi Yan, Jin Zhou, and Zhonghua Cai. 2021. "Assembly of a Benthic Microbial Community in a Eutrophic Bay with a Long History of Oyster Culturing" Microorganisms 9, no. 10: 2019. https://doi.org/10.3390/microorganisms9102019
APA StyleSong, X., Song, J., Yan, Q., Zhou, J., & Cai, Z. (2021). Assembly of a Benthic Microbial Community in a Eutrophic Bay with a Long History of Oyster Culturing. Microorganisms, 9(10), 2019. https://doi.org/10.3390/microorganisms9102019