Grapevine Microbiota Reflect Diversity among Compartments and Complex Interactions within and among Root and Shoot Systems

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sample Collection
2.2. DNA Extraction, Amplification, and Sequencing
2.3. Bioinformatic Processing
2.4. Statistical Analyses
3. Results
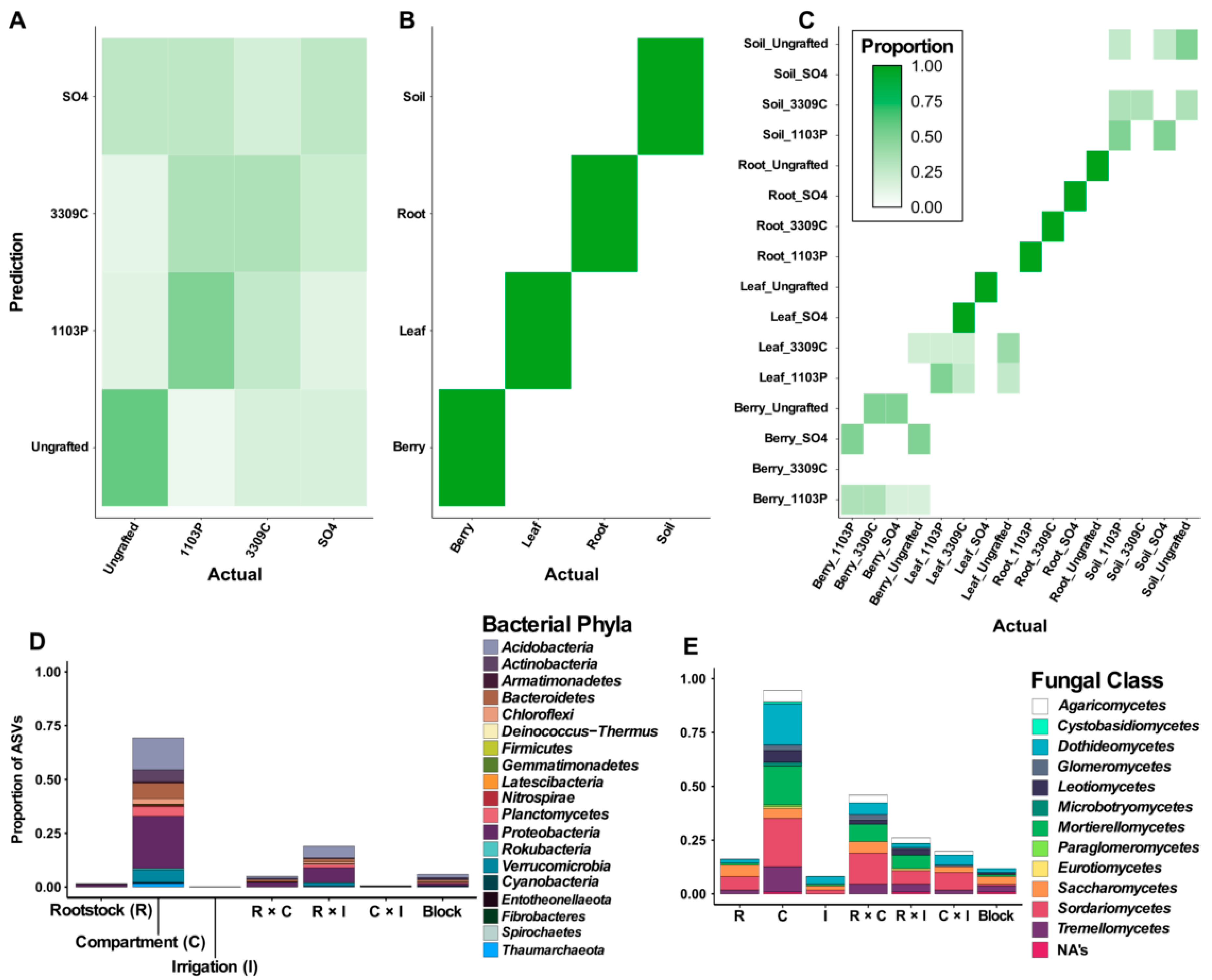
3.1. Bacterial and Fungal Community Composition and Richness Strongly Associate with Plant Compartment
3.2. Rootstock Genotype and Irrigation have Subtle Effects on Community Patterns
3.3. Rootstock and Irrigation Influence Microbiota of Winemaking Relevance
3.4. Machine Learning and Differential Abundance Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chi, F.; Shen, S.-H.; Cheng, H.; Jing, Y.-X.; Yanni, Y.G.; Dazzo, F.B. Ascending Migration of Endophytic Rhizobia, from Roots to Leaves, inside Rice Plants and Assessment of Benefits to Rice Growth Physiology. Appl. Environ. Microbiol. 2005, 71, 7271–7278. [Google Scholar] [CrossRef] [PubMed]
- Martins, G.; Lauga, B.; Miot-Sertier, C.; Mercier, A.; Lonvaud, A.; Soulas, M.-L.; Soulas, G.; Masneuf-Pomarède, I. Characterization of Epiphytic Bacterial Communities from Grapes, Leaves, Bark and Soil of Grapevine Plants Grown, and Their Relations. PLoS ONE 2013, 8, e73013. [Google Scholar] [CrossRef] [PubMed]
- Hacquard, S.; Garrido-Oter, R.; González, A.; Spaepen, S.; Ackermann, G.; Lebeis, S.; McHardy, A.C.; Dangl, J.L.; Knight, R.; Ley, R.; et al. Microbiota and Host Nutrition across Plant and Animal Kingdoms. Cell Host Microbe 2015, 17, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef]
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Themaat, E.V.L.; Schulze-Lefert, P. Structure and Functions of the Bacterial Microbiota of Plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef]
- Lau, J.A.; Lennon, J.T. Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc. Natl. Acad. Sci. USA 2012, 109, 14058–14062. [Google Scholar] [CrossRef]
- Keymer, D.P.; Lankau, R.A. Disruption of plant-soil-microbial relationships influences plant growth. J. Ecol. 2017, 105, 816–827. [Google Scholar] [CrossRef]
- Walters, W.A.; Jin, Z.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q.; et al. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. USA 2018, 115, 7368–7373. [Google Scholar] [CrossRef]
- Coleman-Derr, D.; Desgarennes, D.; Fonseca-Garcia, C.; Gross, S.; Clingenpeel, S.; Woyke, T.; North, G.; Visel, A.; Partida-Martinez, L.P.; Tringe, S.G. Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 2016, 209, 798–811. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Green, J.L.; Holmes, A.J.; Westoby, M.; Oliver, I.; Briscoe, D.; Dangerfield, M.; Gillings, M.; Beattie, A.J. Spatial scaling of microbial eukaryote diversity. Nature 2004, 432, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fire, A.Z.; Boyd, S.; Olshen, R.A. Estimating Clonality. Proc. Natl. Acad. Sci. USA 2014, 110, 6548–6553. [Google Scholar]
- Jiang, Y.; Li, S.; Li, R.; Zhang, J.; Liu, Y.; Lv, L.; Zhu, H.; Wu, W.; Li, W. Plant cultivars imprint the rhizosphere bacterial community composition and association networks. Soil Biol. Biochem. 2017, 109, 145–155. [Google Scholar] [CrossRef]
- Mahoney, A.K.; Yin, C.; Hulbert, S.H. Community Structure, Species Variation, and Potential Functions of Rhizosphere-Associated Bacteria of Different Winter Wheat (Triticum aestivum) Cultivars. Front. Plant Sci. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Fitzpatrick, C.R.; Copeland, J.; Wang, P.W.; Guttman, D.S.; Kotanen, P.M.; Johnson, M.T.J. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc. Natl. Acad. Sci. USA 2018, 115, E1157–E1165. [Google Scholar] [CrossRef]
- Adams, P.D.; Kloepper, J.W. Effect of host genotype on indigenous bacterial endophytes of cotton (Gossypium hirsutum L.). Plant Soil 2002, 240, 181–189. [Google Scholar] [CrossRef]
- Leff, J.W.; Lynch, R.C.; Kane, N.C.; Fierer, N. Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus. New Phytol. 2016, 214, 412–423. [Google Scholar] [CrossRef]
- İnceoğlu, Ã.; Salles, J.F.; van Overbeek, L.; van Elsas, J.D. Effects of Plant Genotype and Growth Stage on the Betaproteobacterial Communities Associated with Different Potato Cultivars in Two Fields. Appl. Environ. Microbiol. 2010, 76, 3675–3684. [Google Scholar] [CrossRef]
- Szoboszlay, M.; Lambers, J.; Chappell, J.; Kupper, J.V.; Moe, L.A.; McNear, D.H. Comparison of root system architecture and rhizosphere microbial communities of Balsas teosinte and domesticated corn cultivars. Soil Biol. Biochem. 2015, 80, 34–44. [Google Scholar] [CrossRef]
- Bouffaud, M.; Kyselková, M.; Gouesnard, B.; Grundmann, G.; Muller, D.; Moënne-loccoz, Y. Is diversification history of maize influencing selection of soil bacteria by roots? Mol. Ecol. 2012, 21, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Micallef, S.A.; Shiaris, M.P.; Colón-Carmona, A. Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J. Exp. Bot. 2009, 60, 1729–1742. [Google Scholar] [CrossRef] [PubMed]
- Sasse, J.; Martinoia, E.; Northen, T. Feed Your Friends: Do Plant Exudates Shape the Root Microbiome? Trends Plant Sci. 2018, 23, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.; Coleman-Derr, D. Drought Stress and Root-Associated Bacterial Communities. Front. Plant Sci. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Rolfe, S.A.; Griffiths, J.; Ton, J. Crying out for help with root exudates: Adaptive mechanisms by which stressed plants assemble health-promoting soil microbiomes. Curr. Opin. Microbiol. 2019, 49, 73–82. [Google Scholar] [CrossRef]
- Yuan, J.; Zhao, J.; Wen, T.; Zhao, M.; Li, R.; Goossens, P.; Huang, Q.; Bai, Y.; Vivanco, J.M.; Kowalchuk, G.A.; et al. Root exudates drive the soil-borne legacy of aboveground pathogen infection. Microbiome 2018, 6, 156. [Google Scholar] [CrossRef]
- Gargallo-Garriga, A.; Preece, C.; Sardans, J.; Oravec, M.; Urban, O.; Peñuelas, J. Root exudate metabolomes change under drought and show limited capacity for recovery. Sci. Rep. 2018, 8, 12696. [Google Scholar] [CrossRef]
- Food and Agriculture Organization. FAOSTAT. 2019. Available online: www.fao.org/faostat/en/#data/QC/Visualize (accessed on 21 January 2019).
- Warschefsky, E.J.; Klein, L.L.; Frank, M.H.; Chitwood, D.H.; Londo, J.P.; von Wettberg, E.J.B.; Miller, A.J. Rootstocks: Diversity, Domestication, and Impacts on Shoot Phenotypes. Trends Plant Sci. 2016, 21, 418–437. [Google Scholar] [CrossRef]
- Wagner, M.R.; Lundberg, D.S.; Del Rio, T.G.; Tringe, S.G.; Dangl, J.L.; Mitchell-Olds, T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Mudge, K.; Janick, J.; Scofield, S.; Goldschmidt, E.E. A History of Grafting. In Horticultural Reviews; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; Volume 35, pp. 437–493. [Google Scholar]
- Pina, A.; Errea, P. A review of new advances in mechanism of graft compatibility–incompatibility. Sci. Hortic. 2005, 106, 1–11. [Google Scholar] [CrossRef]
- Gaut, B.S.; Miller, A.J.; Seymour, D.K. Living with Two Genomes: Grafting and Its Implications for Plant Genome-to-Genome Interactions, Phenotypic Variation, and Evolution. Annu. Rev. Genet. 2019, 53, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Kliman, T. The Wild Vine: A Forgotten Grape and the Untold Story of American Wine; Print.; Broadway Paperbacks: New York, NY, USA, 2010. [Google Scholar]
- Ollat, N.; Bordenave, L.; Tandonnet, J.P.; Boursiquot, J.M.; Marguerit, E. Grapevine rootstocks: Origins and perspectives. Acta Hortic. 2016, 11–22. [Google Scholar] [CrossRef]
- Gautier, A.; Cookson, S.J.; Lagalle, L.; Ollat, N.; Marguerit, E. Influence of the three main genetic backgrounds of grapevine rootstocks on petiolar nutrient concentrations of the scion, with a focus on phosphorus. OENO One 2020, 54, 1–13. [Google Scholar] [CrossRef]
- Lecourt, J.; Lauvergeat, V.; Ollat, N.; Vivin, P.; Cookson, S.J. Shoot and root ionome responses to nitrate supply in grafted grapevines are rootstock genotype dependent. Aust. J. Grape Wine Res. 2015, 21, 311–318. [Google Scholar] [CrossRef]
- Cookson, S.; Ollat, N. Grafting with rootstocks induces extensive transcriptional re-programming in the shoot apical meristem of grapevine. BMC Plant Biol. 2013, 13, 147. [Google Scholar] [CrossRef]
- Migicovsky, Z.; Harris, Z.N.; Klein, L.L.; Li, M.; McDermaid, A.; Chitwood, D.H.; Fennell, A.; Kovacs, L.G.; Kwasniewski, M.; Londo, J.P.; et al. Rootstock effects on scion phenotypes in a ‘Chambourcin’ experimental vineyard. Hortic. Res. 2019, 6, 64. [Google Scholar] [CrossRef]
- D’Amico, F.; Candela, M.; Turroni, S.; Biagi, E.; Brigidi, P.; Bega, A.; Vancini, D.; Rampelli, S. The rootstock regulates microbiome diversity in root and rhizosphere compartments of Vitis vinifera cultivar Lambrusco. Front. Microbiol. 2018, 9, 2240. [Google Scholar] [CrossRef]
- Marasco, R.; Rolli, E.; Fusi, M.; Michoud, G.; Daffonchio, D. Grapevine rootstocks shape underground bacterial microbiome and networking but not potential functionality. Microbiome 2018, 6, 3. [Google Scholar] [CrossRef]
- Berlanas, C.; Berbegal, M.; Elena, G.; Laidani, M.; Cibriain, J.F.; Sagües, A.; Gramaje, D. The Fungal and Bacterial Rhizosphere Microbiome Associated With Grapevine Rootstock Genotypes in Mature and Young Vineyards. Front. Microbiol. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Reisch, B.I.; Owens, C.L.; Cousins, P.S. Grape. In Fruit Breeding; Badenes, M.L., Byrne, D.H., Eds.; Springer: Boston, MA, USA, 2012; pp. 225–262. [Google Scholar]
- Wilcox, W.F.; Gubler, W.D.; Uyemoto, J.K. PART I: Diseases Caused by Biotic Factors. In Compendium of Grape Diseases, Disorders, and Pests, Second Edition; Wilcox, W.F., Gubler, W.D., Uyemoto, J.K., Eds.; The American Phytopathological Society: St. Paul, MN, USA, 2015; pp. 17–146. [Google Scholar]
- Bisiach, M.; Minervini, G.; Salomone, M.C. Experimental research on acid bounch rot and its relationship to gray mold. EPPO Bull. 1982, 12, 15–27. [Google Scholar] [CrossRef]
- Ioriatti, C.; Guzzon, R.; Anfora, G.; Ghidoni, F.; Mazzoni, V.; Villegas, T.R.; Dalton, D.T.; Walton, V.M. Drosophila suzukii (Diptera: Drosophilidae) Contributes to the Development of Sour Rot in Grape. J. Econ. Entomol. 2018, 111, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Barata, A.; Pais, A.; Malfeito-Ferreira, M.; Loureiro, V. Influence of sour rotten grapes on the chemical composition and quality of grape must and wine. Eur. Food Res. Technol. 2011, 233, 183–194. [Google Scholar] [CrossRef]
- Barata, A.; Campo, E.; Malfeito-Ferreira, M.; Loureiro, V.; Cacho, J.; Ferreira, V. Analytical and Sensorial Characterization of the Aroma of Wines Produced with Sour Rotten Grapes Using GC-O and GC-MS: Identification of Key Aroma Compounds. J. Agric. Food Chem. 2011, 59, 2543–2553. [Google Scholar] [CrossRef]
- Zoecklein, B.W.; Williams, J.M.; Duncan, S.E. Effect of Sour Rot on the Composition of White Riesling (Vitis vinifera L.) Grapes. Small Fruits Rev. 2000, 1, 63–77. [Google Scholar] [CrossRef]
- Hall, M.E.; Loeb, G.M.; Cadle-Davidson, L.; Evans, K.J.; Wilcox, W.F. Grape Sour Rot: A Four-Way Interaction Involving the Host, Yeast, Acetic Acid Bacteria, and Insects. Phytopathology 2018, 108, 1429–1442. [Google Scholar] [CrossRef]
- Hall, M.E.; Wilcox, W.F. Identification and Frequencies of Endophytic Microbes within Healthy Grape Berries. Am. J. Enol. Vitic. 2019, 70, 212–219. [Google Scholar] [CrossRef]
- Pinto, C.; Pinho, D.; Sousa, S.; Pinheiro, M.; Egas, C.; Gomes, A.C. Unravelling the Diversity of Grapevine Microbiome. PLoS ONE 2014, 9, e85622. [Google Scholar] [CrossRef]
- Hall, M.E.; O’Bryon, I.; Wilcox, W.F.; Osier, M.V.; Cadle-Davidson, L. The epiphytic microbiota of sour rot-affected grapes differs minimally from that of healthy grapes, indicating causal organisms are already present on healthy berries. PLoS ONE 2019, 14, e0211378. [Google Scholar] [CrossRef]
- Barata, A.; Malfeito-Ferreira, M.; Loureiro, V. The microbial ecology of wine grape berries. Int. J. Food Microbiol. 2012, 153, 243–259. [Google Scholar] [CrossRef]
- Malfeito-Ferreira, M. Spoilage Yeasts in Red Wines. In Red Wine Technology; Morata, A., Ed.; Elsevier: London, UK, 2018; pp. 219–235. [Google Scholar]
- Maimaitiyiming, M.; Ghulam, A.; Bozzolo, A.; Wilkins, J.L.; Kwasniewski, M.T. Early Detection of Plant Physiological Responses to Different Levels of Water Stress Using Reflectance Spectroscopy. Remote Sens. 2017, 9, 745. [Google Scholar] [CrossRef]
- Jones, G.V. Winegrape Phenology. In Phenology: An Integrative Environmental Science; Schwartz, M.D., Ed.; Springer: Dordrecht, The Netherlands, 2003; pp. 523–539. [Google Scholar]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.P.; Peay, K.G. Sequence Depth, Not PCR Replication, Improves Ecological Inference from Next Generation DNA Sequencing. PLoS ONE 2014, 9, e90234. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Fungal Barcoding Consortium Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [PubMed]
- Glassman, S.I.; Martiny, J.B.H. Broadscale Ecological Patterns Are Robust to Use of Exact Sequence Variants versus Operational Taxonomic Units. mSphere 2018, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Unite Community. UNITE Release 18.11.2018. Available online: https://plutof.ut.ee/#/doi/10.15156/BIO/786334.
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2019. Available online: https://www.r-project.org/.
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2016. [Google Scholar]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 3rd ed.; Sage: Thousands Oaks, CA, USA, 2019. [Google Scholar]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Oksanen, J.; Guillaume Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, 2019.
- Lenth, R.; Love, J.; Herve, M. emmeans: Estimated Marginal Means, aka Least-Squares Means. 2020. Available online: https://cran.r-project.org/package=emmeans.
- Wright, M.N.; Ziegler, A. ranger: A Fast Implementation of Random Forests for High Dimensional Data in C++ and R. J. Stat. Softw. 2017, 77. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 159–160. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S.; et al. The Soil Microbiome Influences Grapevine-Associated Microbiota. MBio 2015, 6, e02527-14. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Howell, K. Community succession of the grapevine fungal microbiome in the annual growth cycle. Environ. Microbiol. 2020. [Google Scholar] [CrossRef]
- Liu, H.; Carvalhais, L.C.; Crawford, M.; Singh, E.; Dennis, P.G.; Pieterse, C.M.J.; Schenk, P.M. Inner Plant Values: Diversity, Colonization and Benefits from Endophytic Bacteria. Front. Microbiol. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tkacz, A.; Bestion, E.; Bo, Z.; Hortala, M.; Poole, P.S. Influence of Plant Fraction, Soil, and Plant Species on Microbiota: A Multikingdom Comparison. MBio 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Ottesen, A.R.; Gorham, S.; Reed, E.; Newell, M.J.; Ramachandran, P.; Canida, T.; Allard, M.; Evans, P.; Brown, E.; White, J.R. Using a Control to Better Understand Phyllosphere Microbiota. PLoS ONE 2016, 11, e0163482. [Google Scholar] [CrossRef]
- Bowers, R.M.; McLetchie, S.; Knight, R.; Fierer, N. Spatial variability in airborne bacterial communities across land-use types and their relationship to the bacterial communities of potential source environments. ISME J. 2011, 5, 601–612. [Google Scholar] [CrossRef]
- Šantl-Temkiv, T.; Gosewinkel, U.; Starnawski, P.; Lever, M.; Finster, K. Aeolian dispersal of bacteria in southwest Greenland: Their sources, abundance, diversity and physiological states. FEMS Microbiol. Ecol. 2018, 94, 1–10. [Google Scholar] [CrossRef]
- Smets, W.; Moretti, S.; Denys, S.; Lebeer, S. Airborne bacteria in the atmosphere: Presence, purpose, and potential. Atmos. Environ. 2016, 139, 214–221. [Google Scholar] [CrossRef]
- Lymperopoulou, D.S.; Adams, R.I.; Lindow, S.E. Contribution of Vegetation to the Microbial Composition of Nearby Outdoor Air. Appl. Environ. Microbiol. 2016, 82, 3822–3833. [Google Scholar] [CrossRef]
- Fahlgren, C.; Hagström, A.; Nilsson, D.; Zweifel, U.L. Annual Variations in the Diversity, Viability, and Origin of Airborne Bacteria. Appl. Environ. Microbiol. 2010, 76, 3015–3302. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.M.; McCubbin, I.B.; Hallar, A.G.; Fierer, N. Seasonal variability in airborne bacterial communities at a high-elevation site. Atmos. Environ. 2012, 50, 41–49. [Google Scholar] [CrossRef]
- Bowers, R.M.; Clements, N.; Emerson, J.B.; Wiedinmyer, C.; Hannigan, M.P.; Fierer, N. Seasonal Variability in Bacterial and Fungal Diversity of the Near-Surface Atmosphere. Environ. Sci. Technol. 2013, 47, 12097–12106. [Google Scholar] [CrossRef] [PubMed]
- Hyma, K.E.; Fay, J.C. Mixing of vineyard and oak-tree ecotypes of Saccharomyces cerevisiae in North American vineyards. Mol. Ecol. 2013, 22, 2917–2930. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.; Jiang, Y.; He, S.Y. The role of water in plant-microbe interactions. Plant J. 2018, 93, 771–780. [Google Scholar] [CrossRef]
- Beattie, G.A. Water Relations in the Interaction of Foliar Bacterial Pathogens with Plants. Annu. Rev. Phytopathol. 2011, 49, 533–555. [Google Scholar] [CrossRef]
- Talley, S.M.; Coley, P.D.; Kursar, T.A. The effects of weather on fungal abundance and richness among 25 communities in the Intermountain West. BMC Ecol. 2002, 2. [Google Scholar] [CrossRef]
- Kadivar, H.; Stapleton, A.E. Ultraviolet Radiation Alters Maize Phyllosphere Bacterial Diversity. Microb. Ecol. 2003, 45, 353–361. [Google Scholar] [CrossRef]
- Leveau, J.H.J.; Lindow, S.E. Appetite of an epiphyte: Quantitative monitoring of bacterial sugar consumption in the phyllosphere. Proc. Natl. Acad. Sci. USA 2001, 98, 3446–3453. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Collins, T.S.; Masarweh, C.; Allen, G.; Heymann, H.; Ebeler, S.E.; Mills, D.A. Associations among Wine Grape Microbiome, Metabolome, and Fermentation Behavior Suggest Microbial Contribution to Regional Wine Characteristics. MBio 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Mezzasalma, V.; Sandionigi, A.; Guzzetti, L.; Galimberti, A.; Grando, M.S.; Tardaguila, J.; Labra, M. Geographical and Cultivar Features Differentiate Grape Microbiota in Northern Italy and Spain Vineyards. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Santoni, S.; This, P.; Péros, J.-P. Genotype-Environment Interaction Shapes the Microbial Assemblage in Grapevine’s Phyllosphere and Carposphere: An NGS Approach. Microorganisms 2018, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Chen, X.; Cui, B.; Bai, Z.; Zhuang, G. Variety features differentiate microbiota in the grape leaves. Can. J. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Naylor, D.; Dong, Z.; Simmons, T.; Pierroz, G.; Hixson, K.K.; Kim, Y.-M.; Zink, E.M.; Engbrecht, K.M.; Wang, Y.; et al. Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E4284–E4293. [Google Scholar] [CrossRef] [PubMed]
- Mönchgesang, S.; Strehmel, N.; Schmidt, S.; Westphal, L.; Taruttis, F.; Müller, E.; Herklotz, S.; Neumann, S.; Scheel, D. Natural variation of root exudates in Arabidopsis thaliana-linking metabolomic and genomic data. Sci. Rep. 2016, 6, 29033. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef]
- Sun, H.; Loeb, G.; Walter-Peterson, H.; Martinson, T.; Scott, J.G. Insecticide Resistance in Drosophila melanogaster (Diptera: Drosophilidae) is Associated with Field Control Failure of Sour Rot Disease in a New York Vineyard. J. Econ. Entomol. 2019, 112, 1498–1501. [Google Scholar] [CrossRef]
- Bartowsky, E.J.; Henschke, P.A. Acetic acid bacteria spoilage of bottled red wine—A review. Int. J. Food Microbiol. 2008, 125, 60–70. [Google Scholar] [CrossRef]
- Bartowsky, E.J.; Xia, D.; Gibson, R.L.; Fleet, G.H.; Henschke, P.A. Spoilage of bottled red wine by acetic acid bacteria. Lett. Appl. Microbiol. 2003, 36, 307–314. [Google Scholar] [CrossRef]
- Ciani, M.; Maccarelli, F. Oenological properties of non-Saccharomyces yeasts associated with wine-making. World J. Microbiol. Biotechnol. 1997, 14, 199–203. [Google Scholar] [CrossRef]
- Ciani, M.; Comitini, F. Use of Non-Saccharomyces Yeasts in Red Winemaking. In Red Wine Technology; Morata, A., Ed.; Elsevier: London, UK, 2018; pp. 51–68. [Google Scholar]
- Padilla, B.; Gil, J.V.; Manzanares, P. Past and Future of Non-Saccharomyces Yeasts: From Spoilage Microorganisms to Biotechnological Tools for Improving Wine Aroma Complexity. Front. Microbiol. 2016, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Jolly, N.P.; Varela, C.; Pretorius, I.S. Not your ordinary yeast: Non- Saccharomyces yeasts in wine production uncovered. FEMS Yeast Res. 2014, 14, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Viana, F.; Gil, J.; Genoves, S.; Valles, S.; Manazanares, P. Rational selection of non-Saccharomyces wine yeasts for mixed starters based on ester formation and enological traits. Food Microbiol. 2008, 25, 778–785. [Google Scholar] [CrossRef]
- Moreira, N.; Mendes, F.; Hogg, T.; Vasconcelos, I. Alcohols, esters and heavy sulphur compounds production by pure and mixed cultures of apiculate wine yeasts. Int. J. Food Microbiol. 2005, 103, 285–294. [Google Scholar] [CrossRef]
- Hernández-Orte, P.; Cersosimo, M.; Loscos, N.; Cacho, J.; Garcia-Moruno, E.; Ferreira, V. The development of varietal aroma from non-floral grapes by yeasts of different genera. Food Chem. 2008, 107, 1064–1077. [Google Scholar] [CrossRef]
- Swangkeaw, J.; Vichitphan, S.; Butzke, C.E.; Vichitphan, K. Characterization of β-glucosidases from Hanseniaspora sp. and Pichia anomala with potentially aroma-enhancing capabilities in juice and wine. World J. Microbiol. Biotechnol. 2011, 27, 423–430. [Google Scholar] [CrossRef]
- Rodriguez, M.E.; Lopes, C.A.; Broock, M.; Valles, S.; Ramon, D.; Caballero, A.C. Screening and typing of Patagonian wine yeasts for glycosidase activities. J. Appl. Microbiol. 2004, 96, 84–95. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Unweighted UniFrac | Fungal Bray–Curtis | |||||
|---|---|---|---|---|---|---|
| Factor | Pseudo-F | p-Value | R2 | Pseudo-F | p-Value | R2 |
| Rootstock (R) | F3176 = 1.124 | 0.252 | 0.011 | F3173 = 2.178 | 0.013 | 0.016 |
| Compartment (C) | F3176 = 40.720 | <0.001 | 0.407 | F3173 = 74.320 | <0.001 | 0.550 |
| Irrigation (I) | F2177 = 1.107 | 0.273 | 0.007 | F2174 = 1.203 | 0.247 | 0.006 |
| Block | F2177 = 1.304 | 0.147 | 0.009 | F2174 = 1.402 | 0.151 | 0.007 |
| R × C | F9170 = 1.057 | 0.326 | 0.032 | F9167 = 1.459 | 0.040 | 0.032 |
| R × I | F6173 = 1.058 | 0.324 | 0.021 | F6170 = 1.146 | 0.261 | 0.017 |
| C × I | F6163 = 1.009 | 0.417 | 0.020 | F6170 = 0.936 | 0.526 | 0.014 |
| C × R × I | F18,161 = 0.989 | 0.501 | 0.059 | F18,158 = 0.998 | 0.473 | 0.044 |
| Residual | 0.433 | 0.313 | ||||
| Acetobacterales | Saccharomycetes | |||
|---|---|---|---|---|
| Factor | F Value | p-Value | F Value | p-Value |
| Rootstock (R) | F3128 = 8.329 | <0.001 | F3124 = 4.337 | 0.006 |
| Compartment (C) | F3128 = 0.002 | 1.000 | F3124 = 0.003 | 1.000 |
| Irrigation (I) | F2129 = 0.169 | 0.844 | F2125 = 0.458 | 0.633 |
| Block | F2129 = 0.347 | 0.708 | F2125 = 0.311 | 0.733 |
| R × C | F9122 = 2.111 | 0.033 | F9118 = 0.969 | 0.469 |
| R × I | F6125 = 9.855 | <0.001 | F6121 = 3.139 | 0.007 |
| C × I | F6125 = 0.046 | 1.000 | F6121 = 0.114 | 0.995 |
| C × R × I | F18,113 = 2.454 | 0.002 | F18,109 = 0.787 | 0.713 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swift, J.F.; Hall, M.E.; Harris, Z.N.; Kwasniewski, M.T.; Miller, A.J. Grapevine Microbiota Reflect Diversity among Compartments and Complex Interactions within and among Root and Shoot Systems. Microorganisms 2021, 9, 92. https://doi.org/10.3390/microorganisms9010092
Swift JF, Hall ME, Harris ZN, Kwasniewski MT, Miller AJ. Grapevine Microbiota Reflect Diversity among Compartments and Complex Interactions within and among Root and Shoot Systems. Microorganisms. 2021; 9(1):92. https://doi.org/10.3390/microorganisms9010092
Chicago/Turabian StyleSwift, Joel F., Megan E. Hall, Zachary N. Harris, Misha T. Kwasniewski, and Allison J. Miller. 2021. "Grapevine Microbiota Reflect Diversity among Compartments and Complex Interactions within and among Root and Shoot Systems" Microorganisms 9, no. 1: 92. https://doi.org/10.3390/microorganisms9010092
APA StyleSwift, J. F., Hall, M. E., Harris, Z. N., Kwasniewski, M. T., & Miller, A. J. (2021). Grapevine Microbiota Reflect Diversity among Compartments and Complex Interactions within and among Root and Shoot Systems. Microorganisms, 9(1), 92. https://doi.org/10.3390/microorganisms9010092