Mycobacterium avium Subspecies paratuberculosis Infects and Replicates within Human Monocyte-Derived Dendritic Cells




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell and Culture
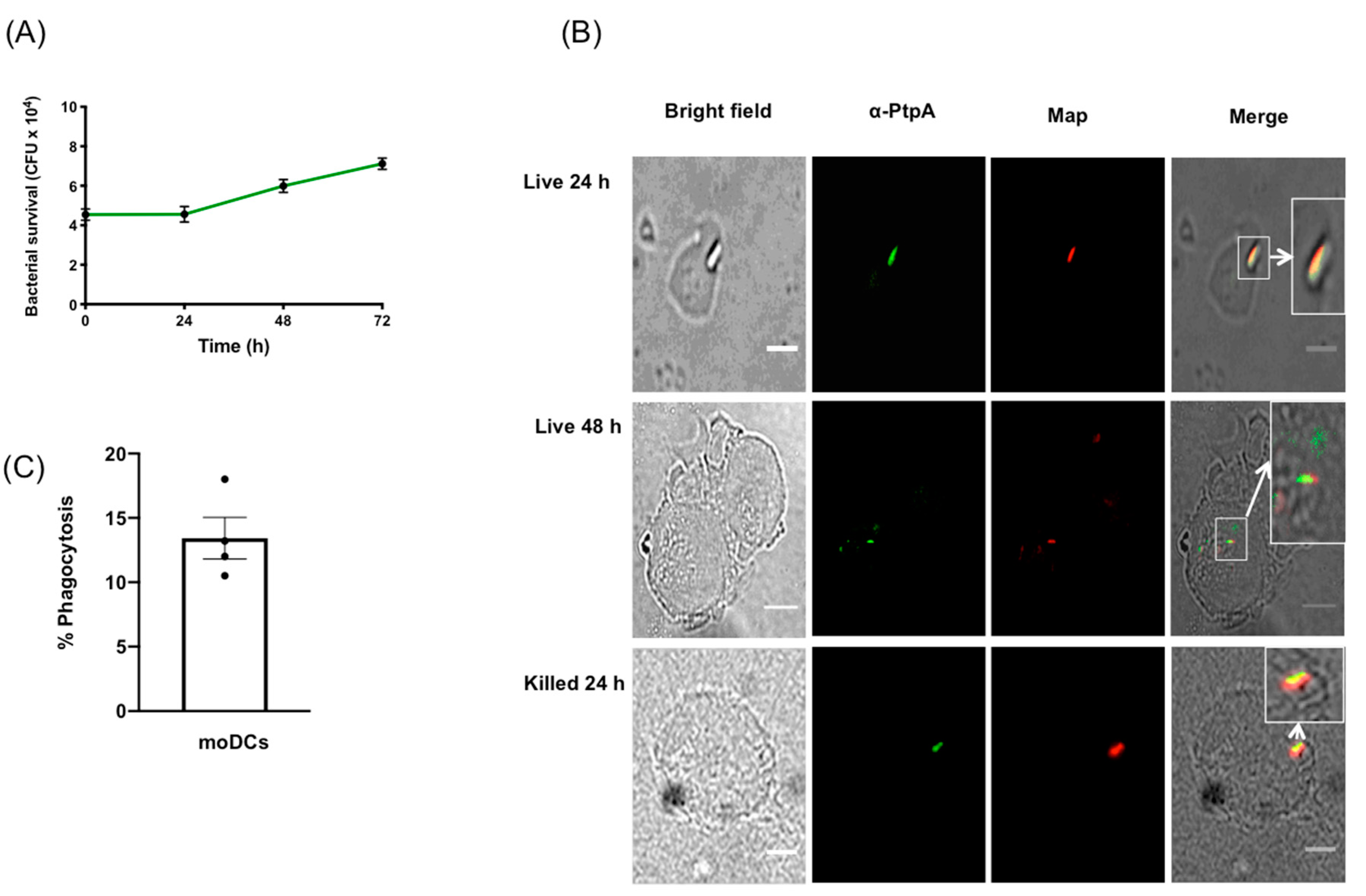
2.2. Phagocytosis Index
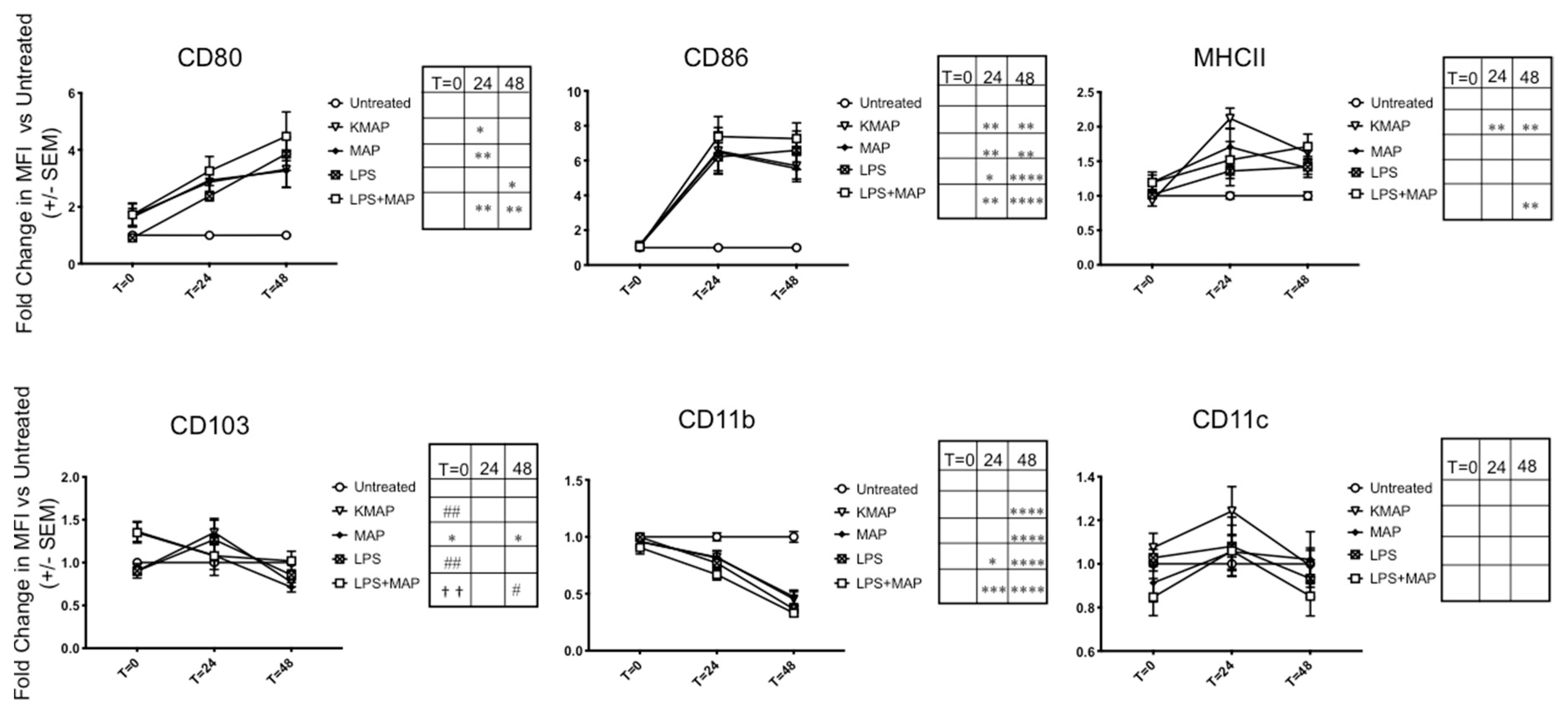
2.3. Flow Cytometry
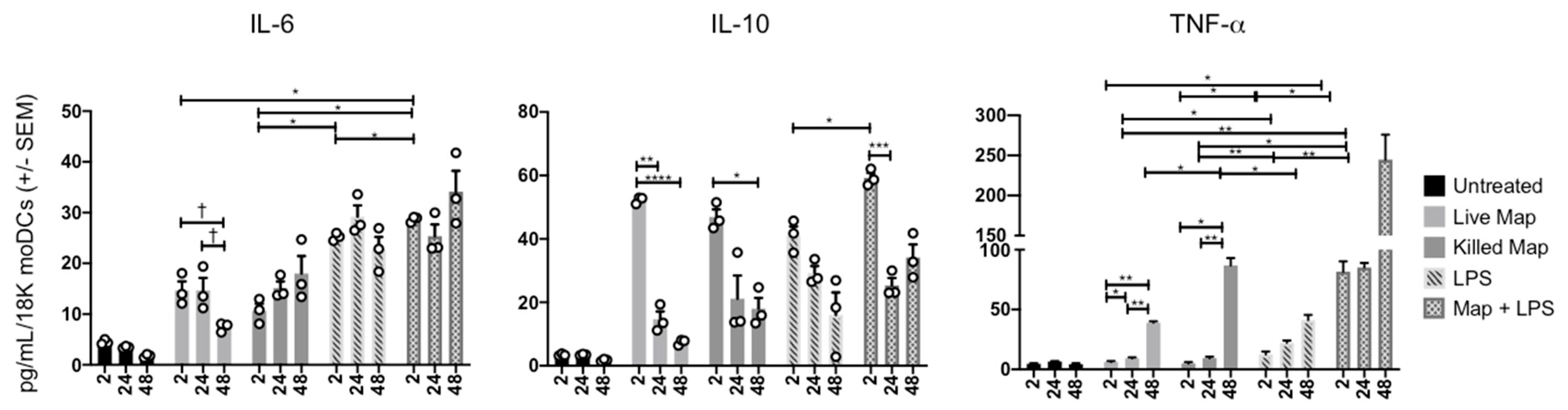
2.4. ELISA
2.5. MAP Colony Counting
2.6. Immunofluorescence
2.7. Statistical Analyses
3. Results
3.1. MAP Infects and Replicates Inside moDCs
3.2. MAP Infection Delays moDC Maturation
3.3. MAP Infection of moDCs Causes Transient Cytokine Production
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chacon, O.; Bermudez, L.E.; Barletta, R.G. Johne’s disease, inflammatory bowel disease, and Mycobacterium paratuberculosis. Annu. Rev. Microbiol. 2004, 58, 329–363. [Google Scholar] [CrossRef]
- Feller, M.; Huwiler, K.; Stephan, R.; Altpeter, E.; Shang, A.; Furrer, H.; Pfyffer, G.E.; Jemmi, T.; Baumgartner, A.; Egger, M. Mycobacterium avium subspecies paratuberculosis and Crohn’s disease: A systematic review and meta-analysis. Lancet Infect. Dis. 2007, 7, 607–613. [Google Scholar] [CrossRef]
- Sanderson, J.D.; Moss, M.T.; Tizard, M.L.; Hermon-Taylor, J. Mycobacterium paratuberculosis DNA in Crohn’s disease tissue. Gut 1992, 33, 890–896. [Google Scholar] [CrossRef][Green Version]
- Scanu, A.M.; Bull, T.J.; Cannas, S.; Sanderson, J.D.; Sechi, L.A.; Dettori, G.; Zanetti, S.; Hermon-Taylor, J. Mycobacterium avium Subspecies paratuberculosis Infection in Cases of Irritable Bowel Syndrome and Comparison with Crohn’s Disease and Johne’s Disease: Common Neural and Immune Pathogenicities. J. Clin. Microbiol. 2007, 45, 3883–3890. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zarei-Kordshouli, F.; Geramizadeh, B.; Khodakaram-Tafti, A. Prevalence of Mycobacterium avium subspecies paratuberculosis IS 900 DNA in biopsy tissues from patients with Crohn’s disease: Histopathological and molecular comparison with Johne’s disease in Fars province of Iran. BMC Infect. Dis. 2019, 19, 23. [Google Scholar] [CrossRef]
- Naser, S.A.; Schwartz, D.; Shafran, I. Isolation of Mycobacterium avium subsp paratuberculosis from breast milk of Crohn’s disease patients. Am. J. Gastroenterol. 2000, 95, 1094–1095. [Google Scholar] [CrossRef]
- Naser, S.A.; Ghobrial, G.; Romero, C.; Valentine, J.F. Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn’s disease. Lancet Lond. Engl. 2004, 364, 1039–1044. [Google Scholar] [CrossRef]
- Naser, S.A.; Collins, M.T.; Crawford, J.T.; Valentine, J.F. Culture of Mycobacterium avium subspecies paratuberculosis (MAP) from the Blood of Patients with Crohn’s disease: A Follow-Up Blind Multi Center Investigation. Open Inflamm. J. 2009, 2, 22–23. [Google Scholar] [CrossRef]
- Zamani, S.; Zali, M.R.; Aghdaei, H.A.; Sechi, L.A.; Niegowska, M.; Caggiu, E.; Keshavarz, R.; Mosavari, N.; Feizabadi, M.M. Mycobacterium avium subsp. paratuberculosis and associated risk factors for inflammatory bowel disease in Iranian patients. Gut Pathog. 2017, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Clancy, R.; Ren, Z.; Turton, J.; Pang, G.; Wettstein, A. Molecular evidence for Mycobacterium avium subspecies paratuberculosis (MAP) in Crohn’s disease correlates with enhanced TNF-alpha secretion. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2007, 39, 445–451. [Google Scholar]
- Nakase, H.; Tamaki, H.; Matsuura, M.; Chiba, T.; Okazaki, K. Involvement of mycobacterium avium subspecies paratuberculosis in TNF-α production from macrophage: Possible link between MAP and immune response in Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, E140–E142. [Google Scholar] [CrossRef] [PubMed]
- Bach, H.; Rosenfeld, G.; Bressler, B. Treatment of Crohn’s disease patients with infliximab is detrimental for the survival of Mycobacterium avium ssp. paratuberculosis within macrophages and shows a remarkable decrease in the immunogenicity of mycobacterial proteins. J. Crohn’s Colitis 2012, 6, 628–629. [Google Scholar] [CrossRef] [PubMed]
- Qasem, A.; Ramesh, S.; Naser, S.A. Genetic polymorphisms in tumour necrosis factor receptors (TNFRSF1A/1B) illustrate differential treatment response to TNFα inhibitors in patients with Crohn’s disease. BMJ Open Gastroenterol. 2019, 6, e000246. [Google Scholar] [CrossRef]
- Bach, H.; Sun, J.; Hmama, Z.; Av-Gay, Y. Mycobacterium avium subsp. paratuberculosis PtpA is an endogenous tyrosine phosphatase secreted during infection. Infect. Immun. 2006, 74, 6540–6546. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bannantine, J.P.; Stabel, J.R. Killing of Mycobacterium avium subspecies paratuberculosis within macrophages. BMC Microbiol. 2002, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Shin, M.-K.; Shin, S.J. MAP1981c, a Putative Nucleic Acid-Binding Protein, Produced by Mycobacterium avium subsp. paratuberculosis, Induces Maturation of Dendritic Cells and Th1-Polarization. Front. Cell. Infect. Microbiol. 2018, 8, 206. [Google Scholar] [CrossRef]
- Kim, W.S.; Kim, J.-S.; Shin, M.-K.; Shin, S.J. A novel Th1-type T-cell immunity-biasing effect of malate dehydrogenase derived from Mycobacterium avium subspecies paratuberculosis via the activation of dendritic cells. Cytokine 2018, 104, 14–22. [Google Scholar] [CrossRef]
- Byun, E.-H.; Kim, W.S.; Kim, J.-S.; Won, C.-J.; Choi, H.-G.; Kim, H.-J.; Cho, S.-N.; Lee, K.; Zhang, T.; Hur, G.M.; et al. Mycobacterium paratuberculosis CobT Activates Dendritic Cells via Engagement of Toll-like Receptor 4 Resulting in Th1 Cell Expansion. J. Biol. Chem. 2012, 287, 38609–38624. [Google Scholar] [CrossRef]
- Basler, T.; Brumshagen, C.; Beineke, A.; Goethe, R.; Bäumer, W. Mycobacterium avium subspecies impair dendritic cell maturation. Innate Immun. 2013, 19, 451–461. [Google Scholar] [CrossRef]
- Bach, H.; Ko, H.H.; Raizman, E.A.; Attarian, R.; Cho, B.; Biet, F.; Enns, R.; Bressler, B. Immunogenicity of Mycobacterium avium subsp. paratuberculosis proteins in Crohn’s disease patients. Scand. J. Gastroenterol. 2011, 46, 30–39. [Google Scholar] [CrossRef]
- Facciuolo, A.; Gonzalez-Cano, P.; Napper, S.; Griebel, P.J.; Mutharia, L.M. Marked Differences in Mucosal Immune Responses Induced in Ileal versus Jejunal Peyer’s Patches to Mycobacterium avium subsp. paratuberculosis Secreted Proteins following Targeted Enteric Infection in Young Calves. PLoS ONE 2016, 11, e0158747. [Google Scholar] [CrossRef] [PubMed]
- Kuehnel, M.P.; Goethe, R.; Habermann, A.; Mueller, E.; Rohde, M.; Griffiths, G.; Valentin-Weigand, P. Characterization of the intracellular survival of Mycobacterium avium ssp. paratuberculosis: Phagosomal pH and fusogenicity in J774 macrophages compared with other mycobacteria. Cell. Microbiol. 2001, 3, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Bach, H.; Papavinasasundaram, K.G.; Wong, D.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe 2008, 3, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Walburger, A.; Koul, A.; Ferrari, G.; Nguyen, L.; Prescianotto-Baschong, C.; Huygen, K.; Klebl, B.; Thompson, C.; Bacher, G.; Pieters, J. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 2004, 304, 1800–1804. [Google Scholar] [CrossRef]
- Nair, S.; Archer, G.E.; Tedder, T.F. Isolation and Generation of Human Dendritic Cells. Curr. Protoc. Immunol. 2012, 99, 7.32.1–7.32.23. [Google Scholar] [CrossRef]
- Bach, H.; Richard-Greenblatt, M.; Bach, E.; Chaffer, M.; Lai, W.; Keefe, G.; Begg, D.J. Protein Kinase G Induces an Immune Response in Cows Exposed to Mycobacterium avium Subsp. paratuberculosis. BioMed Res. Int. 2018, 2018, 1450828. [Google Scholar] [CrossRef]
- Rees, W.D.; Stahl, M.; Jacobson, K.; Bressler, B.; Sly, L.M.; Vallance, B.A.; Steiner, T.S. Enteroids derived from inflammatory bowel disease patients display dysregulated endoplasmic reticulum stress pathways, leading to differential inflammatory responses and dendritic cell maturation. J. Crohn’s Colitis 2019. [Google Scholar] [CrossRef]
- Wong, D.; Bach, H.; Sun, J.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc. Natl. Acad. Sci. USA 2011, 108, 19371–19376. [Google Scholar] [CrossRef]
- Scott, C.L.; Aumeunier, A.M.; Mowat, A.M.C.I. Intestinal CD103+ dendritic cells: Master regulators of tolerance? Trends Immunol. 2011, 32, 412–419. [Google Scholar] [CrossRef]
- Weiss, D.J.; Evanson, O.A.; McClenahan, D.J.; Abrahamsen, M.S.; Walcheck, B.K. Regulation of Expression of Major Histocompatibility Antigens by Bovine Macrophages Infected with Mycobacterium avium subsp. paratuberculosis or Mycobacterium avium subsp. avium. Infect. Immun. 2001, 69, 1002–1008. [Google Scholar] [CrossRef]
- Naser, S.A.; Sagramsingh, S.R.; Naser, A.S.; Thanigachalam, S. Mycobacterium avium subspecies paratuberculosis causes Crohn’s disease in some inflammatory bowel disease patients. World J. Gastroenterol. 2014, 20, 7403–7415. [Google Scholar] [CrossRef]
- Suwandi, A.; Bargen, I.; Pils, M.C.; Krey, M.; Zur Lage, S.; Singh, A.K.; Basler, T.; Falk, C.S.; Seidler, U.; Hornef, M.W.; et al. CD4 T Cell Dependent Colitis Exacerbation Following Re-Exposure of Mycobacterium avium ssp. paratuberculosis. Front. Cell. Infect. Microbiol. 2017, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Suwandi, A.; Bargen, I.; Roy, B.; Pils, M.C.; Krey, M.; Zur Lage, S.; Basler, T.; Rohde, M.; Falk, C.S.; Hornef, M.W.; et al. Experimental colitis is exacerbated by concomitant infection with Mycobacterium avium ssp. paratuberculosis. Inflamm. Bowel Dis. 2014, 20, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Whittington, R.J.; Marshall, D.J.; Nicholls, P.J.; Marsh, I.B.; Reddacliff, L.A. Survival and Dormancy of Mycobacterium avium subsp. paratuberculosis in the Environment. Appl. Environ. Microbiol. 2004, 70, 2989–3004. [Google Scholar] [CrossRef] [PubMed]
- Whittington, R.J.; Marsh, I.B.; Reddacliff, L.A. Survival of Mycobacterium avium subsp. paratuberculosis in Dam Water and Sediment. Appl. Environ. Microbiol. 2005, 71, 5304–5308. [Google Scholar] [CrossRef]
- Ellingson, J.L.E.; Anderson, J.L.; Koziczkowski, J.J.; Radcliff, R.P.; Sloan, S.J.; Allen, S.E.; Sullivan, N.M. Detection of Viable Mycobacterium avium subsp. paratuberculosis in Retail Pasteurized Whole Milk by Two Culture Methods and PCR. J. Food Prot. 2005, 68, 966–972. [Google Scholar] [CrossRef]
- Grant, I.R.; Ball, H.J.; Rowe, M.T. Incidence of Mycobacterium paratuberculosis in Bulk Raw and Commercially Pasteurized Cows’ Milk from Approved Dairy Processing Establishments in the United Kingdom. Appl. Environ. Microbiol. 2002, 68, 2428–2435. [Google Scholar] [CrossRef]
- Spahr, U.; Schafroth, K. Fate of Mycobacterium avium subsp.paratuberculosis in Swiss Hard and Semihard Cheese Manufactured from Raw Milk. Appl. Environ. Microbiol. 2001, 67, 4199–4205. [Google Scholar] [CrossRef]
- Donaghy, J.A.; Totton, N.L.; Rowe, M.T. Persistence of Mycobacterium paratuberculosis during Manufacture and Ripening of Cheddar Cheese. Appl. Environ. Microbiol. 2004, 70, 4899–4905. [Google Scholar] [CrossRef]
- Tailleux, L.; Schwartz, O.; Herrmann, J.-L.; Pivert, E.; Jackson, M.; Amara, A.; Legres, L.; Dreher, D.; Nicod, L.P.; Gluckman, J.C.; et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 2003, 197, 121–127. [Google Scholar] [CrossRef]
- Roberts, L.L.; Robinson, C.M. Mycobacterium tuberculosis infection of human dendritic cells decreases integrin expression, adhesion and migration to chemokines. Immunology 2014, 141, 39–51. [Google Scholar] [CrossRef]
- Kabara, E.; Coussens, P.M. Infection of Primary Bovine Macrophages with Mycobacterium avium Subspecies paratuberculosis Suppresses Host Cell Apoptosis. Front. Microbiol. 2012, 3, 215. [Google Scholar] [CrossRef]
- Hostetter, J.; Kagan, R.; Steadham, E. Opsonization Effects on Mycobacterium avium subsp. paratuberculosis-Macrophage Interactions. Clin. Diagn. Lab. Immunol. 2005, 12, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Mathie, H.A.; Jensen, K.; Stevens, J.M.; Glass, E.J.; Hope, J.C. Quantifying Mycobacterium avium subspecies paratuberculosis infection of bovine monocyte derived macrophages by confocal microscopy. J. Microbiol. Methods 2020, 168, 105779. [Google Scholar] [CrossRef] [PubMed]
- Gorvel, L.; Textoris, J.; Banchereau, R.; Amara, A.B.; Tantibhedhyangkul, W.; Bargen, K.; von Ka, M.B.; Capo, C.; Ghigo, E.; Gorvel, J.-P.; et al. Intracellular Bacteria Interfere with Dendritic Cell Functions: Role of the Type I Interferon Pathway. PLoS ONE 2014, 9, e99420. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Gómez, I.M.; Käser, T.; Gómez-Laguna, J.; Lamp, B.; Sinn, L.; Rümenapf, T.; Carrasco, L.; Saalmüller, A.; Gerner, W. PRRSV-infected monocyte-derived dendritic cells express high levels of SLA-DR and CD80/86 but do not stimulate PRRSV-naïve regulatory T cells to proliferate. Vet. Res. 2015, 46, 54. [Google Scholar] [CrossRef] [PubMed]
- Sebrell, T.A.; Hashimi, M.; Sidar, B.; Wilkinson, R.A.; Kirpotina, L.; Quinn, M.T.; Malkoç, Z.; Taylor, P.J.; Wilking, J.N.; Bimczok, D. A Novel Gastric Spheroid Co-culture Model Reveals Chemokine-Dependent Recruitment of Human Dendritic Cells to the Gastric Epithelium. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 157–171.e3. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rees, W.D.; Lorenzo-Leal, A.C.; Steiner, T.S.; Bach, H. Mycobacterium avium Subspecies paratuberculosis Infects and Replicates within Human Monocyte-Derived Dendritic Cells. Microorganisms 2020, 8, 994. https://doi.org/10.3390/microorganisms8070994
Rees WD, Lorenzo-Leal AC, Steiner TS, Bach H. Mycobacterium avium Subspecies paratuberculosis Infects and Replicates within Human Monocyte-Derived Dendritic Cells. Microorganisms. 2020; 8(7):994. https://doi.org/10.3390/microorganisms8070994
Chicago/Turabian StyleRees, William D., Ana C. Lorenzo-Leal, Theodore S. Steiner, and Horacio Bach. 2020. "Mycobacterium avium Subspecies paratuberculosis Infects and Replicates within Human Monocyte-Derived Dendritic Cells" Microorganisms 8, no. 7: 994. https://doi.org/10.3390/microorganisms8070994
APA StyleRees, W. D., Lorenzo-Leal, A. C., Steiner, T. S., & Bach, H. (2020). Mycobacterium avium Subspecies paratuberculosis Infects and Replicates within Human Monocyte-Derived Dendritic Cells. Microorganisms, 8(7), 994. https://doi.org/10.3390/microorganisms8070994