Bile Acid Sequestrant, Sevelamer Ameliorates Hepatic Fibrosis with Reduced Overload of Endogenous Lipopolysaccharide in Experimental Nonalcoholic Steatohepatitis

 , , ,
, , ,  and
and
Abstract
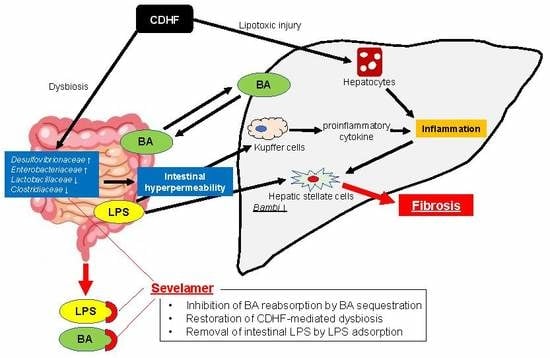
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Protocol
2.2. Cell Culture
2.3. Histological and Immunohistochemical Analyses
2.4. Measurement of Total Bile Acid Levels
2.5. Measurement of LPS Levels
2.6. RNA Extraction and Reverse Transcription-Quantitative Polymerase Chain Reaction
2.7. Protein Extraction and Western Blotting
2.8. Analysis of Gut Microbiota
2.9. Statistical Analyses
3. Results
3.1. Sevelamer Improves CDHF Diet-Induced Steatohepatitis
3.2. Sevelamer Decreases Hepatic Macrophage Infiltration, Inflammatory Response, and Fibrosis in CDHF Diet-Fed Mice
3.3. Sevelamer Reduces Portal Bile Acid Levels and Inhibits the Activation of Hepatic and Intestinal FXR Signaling in CDHF Diet-Fed Mice
3.4. Sevelamer Recovers the Impaired Gut Microbiota and Intestinal Barrier Function in CDHF Diet-Fed Mice
3.5. Sevelamer-Mediated Reduction in LPS Contributes to Suppressing HSC Activation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med.. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.F.; Caussy, C.; Loomba, R. Combination Therapy for Non-Alcoholic Steatohepatitis: Rationale, Opportunities and Challenges. Gut 2020, gutjnl-2019-319104. [Google Scholar] [CrossRef]
- Cardoso, A.C.; de Figueiredo-Mendes, C.; Villela-Nogueira, C.A.; Sanyal, A.J. New Drugs for Non-Alcoholic Steatohepatitis. Liver Int. 2020, 40, 96–101. [Google Scholar] [CrossRef]
- Italian Association for the Study of the Liver (AISF). AISF Position Paper on Nonalcoholic Fatty Liver Disease (NAFLD): Updates and Future Directions. Dig Liver Dis. 2017, 49, 471–483. [Google Scholar] [CrossRef]
- Nakanishi, K.; Kaji, K.; Kitade, M.; Kubo, T.; Furukawa, M.; Saikawa, S.; Shimozato, N.; Sato, S.; Seki, K.; Kawaratani, H.; et al. Exogenous Administration of Low-Dose Lipopolysaccharide Potentiates Liver Fibrosis in a Choline-Deficient l-Amino-Acid-Defined Diet-Induced Murine Steatohepatitis Model. Int. J. Mol. Sci. 2019, 20, E2724. [Google Scholar] [CrossRef]
- Gäbele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Schölmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef]
- Ceccarelli, S.; Panera, N.; Mina, M.; Gnani, D.; De, S.C.; Crudele, A.; Rychlicki, C.; Petrini, S.; Bruscalupi, G.; Agostinelli, L.; et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 2015, 8, 41434–41452. [Google Scholar] [CrossRef] [PubMed]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Douhara, A.; Moriya, K.; Yoshiji, H.; Noguchi, R.; Namisaki, T.; Kitade, M.; Kaji, K.; Aihara, Y.; Nishimura, N.; Takeda, K.; et al. Reduction of endotoxin attenuates liver fibrosis through suppression of hepatic stellate cell activation and remission of intestinal permeability in a rat non-alcoholic steatohepatitis model. Mol. Med. Rep. 2015, 11, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Seki, K.; Kitade, M.; Nishimura, N.; Kaji, K.; Asada, K.; Namisaki, T.; Moriya, K.; Kawaratani, H.; Okura, Y.; Takaya, H.; et al. Oral administration of fructose exacerbates liver fibrosis and hepatocarcinogenesis via increased intestinal permeability in a rat steatohepatitis model. Oncotarget 2018, 9, 28638–28651. [Google Scholar] [CrossRef] [PubMed]
- Stacey, M.; Webb, M. Studies on the antibacterial properties of the bile acids and some compounds derived from cholanic acid. Proc. R. Soc. Med. 1947, 134, 523–537. [Google Scholar]
- Lorenzo-Zúñiga, V.; Bartolí, R.; Planas, R.; Hofmann, A.F.; Viñado, B.; Hagey, L.R.; Hernández, J.M.; Mañé, J.; Alvarez, M.A.; Ausina, V.; et al. Oral Bile Acids Reduce Bacterial Overgrowth, Bacterial Translocation, and Endotoxemia in Cirrhotic Rats. Hepatology 2003, 37, 551–557. [Google Scholar] [CrossRef]
- Ogata, Y.; Nishi, M.; Nakayama, H.; Kuwahara, T.; Ohnishi, Y.; Tashiro, S. Role of Bile in Intestinal Barrier Function and Its Inhibitory Effect on Bacterial Translocation in Obstructive Jaundice in Rats. J. Surg. Res. 2003, 115, 18–23. [Google Scholar] [CrossRef]
- Jamar, G.; Estadella, D.; Pisani, L.P. Contribution of Anthocyanin-Rich Foods in Obesity Control Through Gut Microbiota Interactions. Biofactors 2017, 43, 507–516. [Google Scholar] [CrossRef]
- Bell, D.S.; O’Keefe, J.H. Rediscovering bile acid sequestrants. Diabetes Obes. Metab. 2009, 11, 1114–1121. [Google Scholar] [CrossRef]
- Davidson, M.H.; Dillon, M.A.; Gordon, B.; Jones, P.; Weiss, S.; Isaacsohn, J.; Toth, P.; Burke, S.K. Colesevelam hydrochloride (cholestagel): A new, potent bile acid sequestrant associated with a low incidence of gastrointestinal side effects. Arch. Intern. Med. 1999, 159, 1893–1900. [Google Scholar] [CrossRef]
- Nerild, H.H.; Christensen, M.B.; Knop, F.K.; Brønden, A. Preclinical discovery and development of colesevelam for the treatment of type 2 diabetes. Expert Opin. Drug Discov. 2018, 13, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.R.; Haeusler, R.A. Bile acids in glucose metabolism and insulin signalling - mechanisms and research needs. Nat. Rev. Endocrinol. 2019, 15, 701–712. [Google Scholar] [CrossRef]
- Sonne, D.P.; Hansen, M.; Knop, F.K. Bile acid sequestrants in type 2 diabetes: Potential effects on GLP1 secretion. Eur. J. Endocrinol. 2014, 171, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Potts, A.; He, T.; Duarte, J.A.; Taussig, R.; Mangelsdorf, D.J.; Kliewer, S.A.; Burgess, S.C. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am. J. Physiol. Gastrointest Liver Physiol. 2013, 304, G371–G380. [Google Scholar] [CrossRef] [PubMed]
- Herrema, H.; Meissner, M.; van Dijk, T.H.; Brufau, G.; Boverhof, R.; Oosterveer, M.H.; Reijngoud, D.J.; Müller, M.; Stellaard, F.; Groen, A.K.; et al. Bile salt sequestration induces hepatic de novo lipogenesis through farnesoid X receptor- and liver X receptor alpha-controlled metabolic pathways in mice. Hepatology 2010, 51, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Taniai, M.; Hashimoto, E.; Tobari, M.; Yatsuji, S.; Haruta, I.; Tokushige, K.; Shiratori, K. Treatment of nonalcoholic steatohepatitis with colestimide. Hepatol. Res. 2009, 39, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Ikegami, H.; Fujisawa, T.; Nojima, K.; Kawabata, Y.; Noso, S.; Babaya, N.; Itoi-Babaya, M.; Yamaji, K.; Hiromine, Y.; et al. Prevention and treatment of obesity, insulin resistance, and diabetes by bile acid-binding resin. Diabetes 2007, 56, 239–247. [Google Scholar] [CrossRef]
- Slatopolsky, E.A.; Burke, S.K.; Dillon, M.A. RenaGel, a nonabsorbed calcium- and aluminum-free phosphate binder, lowers serum phosphorus and parathyroid hormone. The RenaGel Study Group. Kidney Int. 1999, 55, 299–307. [Google Scholar] [CrossRef]
- McGettigan, B.M.; McMahan, R.H.; Luo, Y.; Wang, X.X.; Orlicky, D.J.; Porsche, C.; Levi, M.; Rosen, H.R. Sevelamer Improves Steatohepatitis, Inhibits Liver and Intestinal Farnesoid X Receptor (FXR), and Reverses Innate Immune Dysregulation in a Mouse Model of Non-alcoholic Fatty Liver Disease. J. Biol. Chem. 2016, 291, 23058–23067. [Google Scholar] [CrossRef]
- Hauser, A.B.; Azevedo, I.R.; Gonçalves, S.; Stinghen, A.; Aita, C.; Pecoits-Filho, R. Sevelamer carbonate reduces inflammation and endotoxemia in an animal model of uremia. Blood Purif 2010, 30, 153–158. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Mora-Fernández, C.; Muros de Fuentes, M.; Donate-Correa, J.; Cazaña-Pérez, V.; García-Pérez, J. Effect of phosphate binders on serum inflammatory profile, soluble CD14, and endotoxin levels in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2011, 6, 2272–2279. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van, N.M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Knight, R. Species Divergence and the Measurement of Microbial Diversity. FEMS Microbial Rev. 2008, 32, 557–578. [Google Scholar] [CrossRef]
- Takahashi, S.; Luo, Y.; Ranjit, S.; Xie, C.; Libby, A.E.; Orlicky, D.J.; Dvornikov, A.; Wang, X.X.; Myakala, K.; Jones, B.A.; et al. Bile Acid Sequestration Reverses Liver Injury and Prevents Progression of NASH in Western Diet-Fed Mice. J. Biol. Chem. 2020, 295, 4733–4747. [Google Scholar] [CrossRef]
- Gupta, B.; Liu, Y.; Chopyk, D.M.; Rai, R.P.; Desai, C.; Kumar, P.; Farris, A.B.; Nusrat, A.; Parkos, C.A.; Anania, F.A.; et al. Western diet-induced increase in colonic bile acids compromises epithelial barrier in nonalcoholic steatohepatitis. FASEB J. 2020, 34, 7089–7102. [Google Scholar] [CrossRef]
- Ishioka, M.; Miura, K.; Minami, S.; Shimura, Y.; Ohnishi, H. Altered Gut Microbiota Composition and Immune Response in Experimental Steatohepatitis Mouse Models. Dig. Dis. Sci. 2017, 62, 396–406. [Google Scholar] [CrossRef]
- Seki, E.; De, M.S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van, N.M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Verbeke, L.; Mannaerts, I.; Schierwagen, R.; Govaere, O.; Klein, S.; Vander, E.I.; Windmolders, P.; Farre, R.; Wenes, M.; Mazzone, M.; et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci. Rep. 2016, 6, 33453. [Google Scholar] [CrossRef]
- Goto, T.; Itoh, M.; Suganami, T.; Kanai, S.; Shirakawa, I.; Sakai, T.; Asakawa, M.; Yoneyama, T.; Kai, T.; Ogawa, Y. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci. Rep. 2018, 8, 8157. [Google Scholar] [CrossRef]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; van de Peppel, I.P.; Gumber, S.; Karpen, S.J.; Dawson, P.A. Attenuation of the Hepatoprotective Effects of Ileal Apical Sodium Dependent Bile Acid Transporter (ASBT) Inhibition in Choline-Deficient L-Amino Acid-Defined (CDAA) Diet-Fed Mice. Front Med. (Lausanne) 2020, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Huang, Y.; Wang, Y.; Wang, P.; Song, H.; Wang, F. Antibiotics induced intestinal tight junction barrier dysfunction is associated with microbiota dysbiosis, activated NLRP3 inflammasome and autophagy. PLoS ONE 2019, 14, e0218384. [Google Scholar] [CrossRef]
- Li, S.; Qi, C.; Zhu, H.; Yu, R.; Xie, C.; Peng, Y.; Yin, S.W.; Fan, J.; Zhao, S.; Sun, J. Lactobacillus reuteri improves gut barrier function and affects diurnal variation of the gut microbiota in mice fed a high-fat diet. Food Funct. 2019, 10, 4705–4715. [Google Scholar] [CrossRef] [PubMed]
- Perianayagam, M.C.; Jaber, B.L. Endotoxin-binding affinity of sevelamer hydrochloride. Am. J. Nephrol. 2008, 28, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Kristoff, J.; Haret-Richter, G.; Ma, D.; Ribeiro, R.M.; Xu, C.; Cornell, E.; Stock, J.L.; He, T.; Mobley, A.D.; Ross, S.; et al. Early microbial translocation blockade reduces SIV-mediated inflammation and viral replication. J. Clin. Investig. 2014, 124, 2802–2806. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef]
- Sharma, A.; Verma, A.K.; Kofron, M.; Kudira, R.; Miethke, A.; Wu, T.; Wang, J.; Gandhi, C.R. Lipopolysaccharide Reverses Hepatic Stellate Cell Activation via Modulation of cMyb, SMADs and C/EBP Transcription Factors. Hepatology 2020. [Google Scholar] [CrossRef]
- Brønden, A.; Mikkelsen, K.; Sonne, D.P.; Hansen, M.; Våben, C.; Gabe, M.N.; Rosenkilde, M.; Tremaroli, V.; Wu, H.; Bäckhed, F.; et al. Glucose-lowering Effects and Mechanisms of the Bile Acid-Sequestering Resin Sevelamer. Diabetes Obes. Metab. 2018, 20, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, M.; Patel, P.; Dunn-Valadez, S.; Dao, C.; Khan, V.; Ali, H.; El-Serag, L.; Hernaez, R.; Sisson, A.; Thrift, A.P.; et al. Women Have Lower Risk of Nonalcoholic Fatty Liver Disease but Higher Risk of Progression vs. Men: A Systematic Review and Meta-analysis. Clin. Gastroenterol Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Guerrerio, A.L.; Colvin, R.M.; Schwartz, A.K.; Molleston, J.P.; Murray, K.F.; Diehl, A.; Mohan, P.; Schwimmer, J.B.; Lavine, J.E.; Torbenson, M.S.; et al. Choline Intake in a Large Cohort of Patients With Nonalcoholic Fatty Liver Disease. Am. J. Clin. Nutr. 2012, 95, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Meneses, G.; Rosetti, M.; Espinosa, A.; Florentino, A.; Bautista, M.; Díaz, G.; Olvera, G.; Bárcena, B.; Fleury, A.; Adalid-Peralta, L.; et al. Recovery From an Acute Systemic and Central LPS-inflammation Challenge Is Affected by Mouse Sex and Genetic Background. PLoS ONE 2018, 13, e0201375. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense (5′–3′) | Antisense (5′–3′) |
|---|---|---|
| Mouse | ||
| Srebf1 | GGAGCCATGGATTGCACATT | GGCCCGGGAAGTCACTGT |
| Mlxipl | CTGGGGACCTAAACAGGAGC | GAAGCCACCCTATAGCTCCC |
| Fas | TGGGTTCTAGCCAGCAGAGT | ACCACCAGAGACCGTTATGC |
| Acc1 | GCCTCTTCCTGACAAACGAG | TGACTGCCGAAACATCTCTG |
| Ppara | GAGGGTTGAGCTCAGTCAGG | GGTCACCTACGAGTGGCATT |
| Pparg | CTGTGAGACCAACAGCCTGA | AATGCGAGTGGTCTTCCATC |
| Tnfa | ACGGCATGGATCTCAAAGAC | AGATAGCAAATCGGCTGACG |
| Il6 | GTTCTCTGGGAAATCGTGGA | TGTACTCCAGGTAGCTATGG |
| Ccl2 | AGGTCCCTGTCATGCTTCTG | TCTGGACCCATTCCTTCTTG |
| Col1a1 | GAGCGGAGAGTACTGGATCG | GCTTCTTTTCCTTGGGGTTC |
| Acta2 | CTGACAGAGGCACCACTGAA | CATCTCCAGAGTCCAGCACA |
| Tgfb1 | TTGCTTCAGCTCCACAGAGA | TGGTTGTAGAGGGCAAGGAC |
| Shp | CGATCCTCTTCAACCCAGATG | AGGGCTCCAAGACTTCACACA |
| Cyp7a1 | AGCAACTAAACAACCTGCCAGTACTA | GTCCGGATATTCAAGGATGCA |
| Fxr | GTTGCCGTGAGGAAGCTAAG | GAACTTGAGGAAACGGGACA |
| Fgf15 | GGCAAGATATACGGGCTGAT | GATGGTGCTTCATGGATCTG |
| Slc10a2 | TGGGTTTCTTCCTGGCTAGACT | TGTTCTGCATTCCAGTTTCCAA |
| Lbp | GGCTGCTGAATCTCTTCCAC | GAGCGGTGATTCCGATTAAA |
| Cd14 | GTCAGGAACTCTGGCTTTGC | TGGCTTTTACCCACTGAACC |
| Tlr4 | GGCAGCAGGTGGAATTGTAT | AGGCCCCAGAGTTTTGTTCT |
| 18S rRNA | TTGACGGAAGGGCACCACCAG | GCACCACCACCCACGGAATCG |
| Human | ||
| BAMBI | GGCAGCATCACAGTAGCATC | GATCGCCACTCCAGCTACAT |
| COL1A1 | CCAAATCTGTCTCCCCAGAA | TCAAAAACGAAGGGGAGATG |
| ACTA2 | GAGACCCTGTTCCAGCCATC | TACATAGTGGTGCCCCCTGA |
| 18S rRNA | AAACGGCTACCACATCCAAG | CCTCCAATGGATCCTCGTTA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuji, Y.; Kaji, K.; Kitade, M.; Kaya, D.; Kitagawa, K.; Ozutsumi, T.; Fujinaga, Y.; Takaya, H.; Kawaratani, H.; Namisaki, T.; et al. Bile Acid Sequestrant, Sevelamer Ameliorates Hepatic Fibrosis with Reduced Overload of Endogenous Lipopolysaccharide in Experimental Nonalcoholic Steatohepatitis. Microorganisms 2020, 8, 925. https://doi.org/10.3390/microorganisms8060925
Tsuji Y, Kaji K, Kitade M, Kaya D, Kitagawa K, Ozutsumi T, Fujinaga Y, Takaya H, Kawaratani H, Namisaki T, et al. Bile Acid Sequestrant, Sevelamer Ameliorates Hepatic Fibrosis with Reduced Overload of Endogenous Lipopolysaccharide in Experimental Nonalcoholic Steatohepatitis. Microorganisms. 2020; 8(6):925. https://doi.org/10.3390/microorganisms8060925
Chicago/Turabian StyleTsuji, Yuki, Kosuke Kaji, Mitsuteru Kitade, Daisuke Kaya, Koh Kitagawa, Takahiro Ozutsumi, Yukihisa Fujinaga, Hiroaki Takaya, Hideto Kawaratani, Tadashi Namisaki, and et al. 2020. "Bile Acid Sequestrant, Sevelamer Ameliorates Hepatic Fibrosis with Reduced Overload of Endogenous Lipopolysaccharide in Experimental Nonalcoholic Steatohepatitis" Microorganisms 8, no. 6: 925. https://doi.org/10.3390/microorganisms8060925
APA StyleTsuji, Y., Kaji, K., Kitade, M., Kaya, D., Kitagawa, K., Ozutsumi, T., Fujinaga, Y., Takaya, H., Kawaratani, H., Namisaki, T., Moriya, K., Akahane, T., & Yoshiji, H. (2020). Bile Acid Sequestrant, Sevelamer Ameliorates Hepatic Fibrosis with Reduced Overload of Endogenous Lipopolysaccharide in Experimental Nonalcoholic Steatohepatitis. Microorganisms, 8(6), 925. https://doi.org/10.3390/microorganisms8060925