An Optimized Metabarcoding Method for Mimiviridae

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Seawater Sampling, Storage, and DNA Extraction
2.2. PCR Conditions, Amplicon Purification Protocols, and Sequencing
2.3. Computational Quality Control of Reads
2.4. qPCR Primer Design and Experiments
2.5. Metabarcoding Analysis of Eukaryotes
2.6. Data and software availability
3. Results
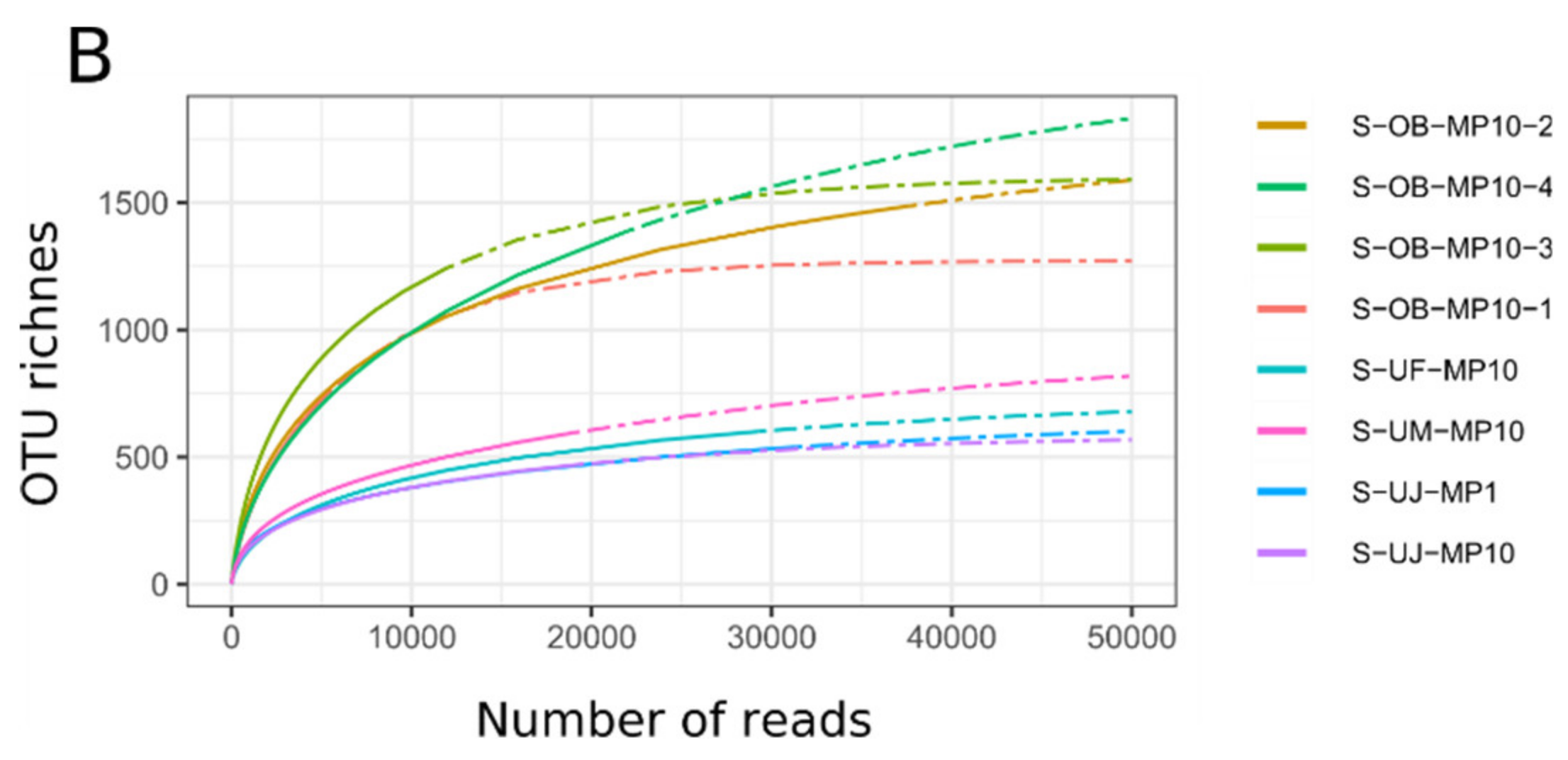
3.1. Mimiviridae Community Profiles were Coherent across Different Primer Cocktails
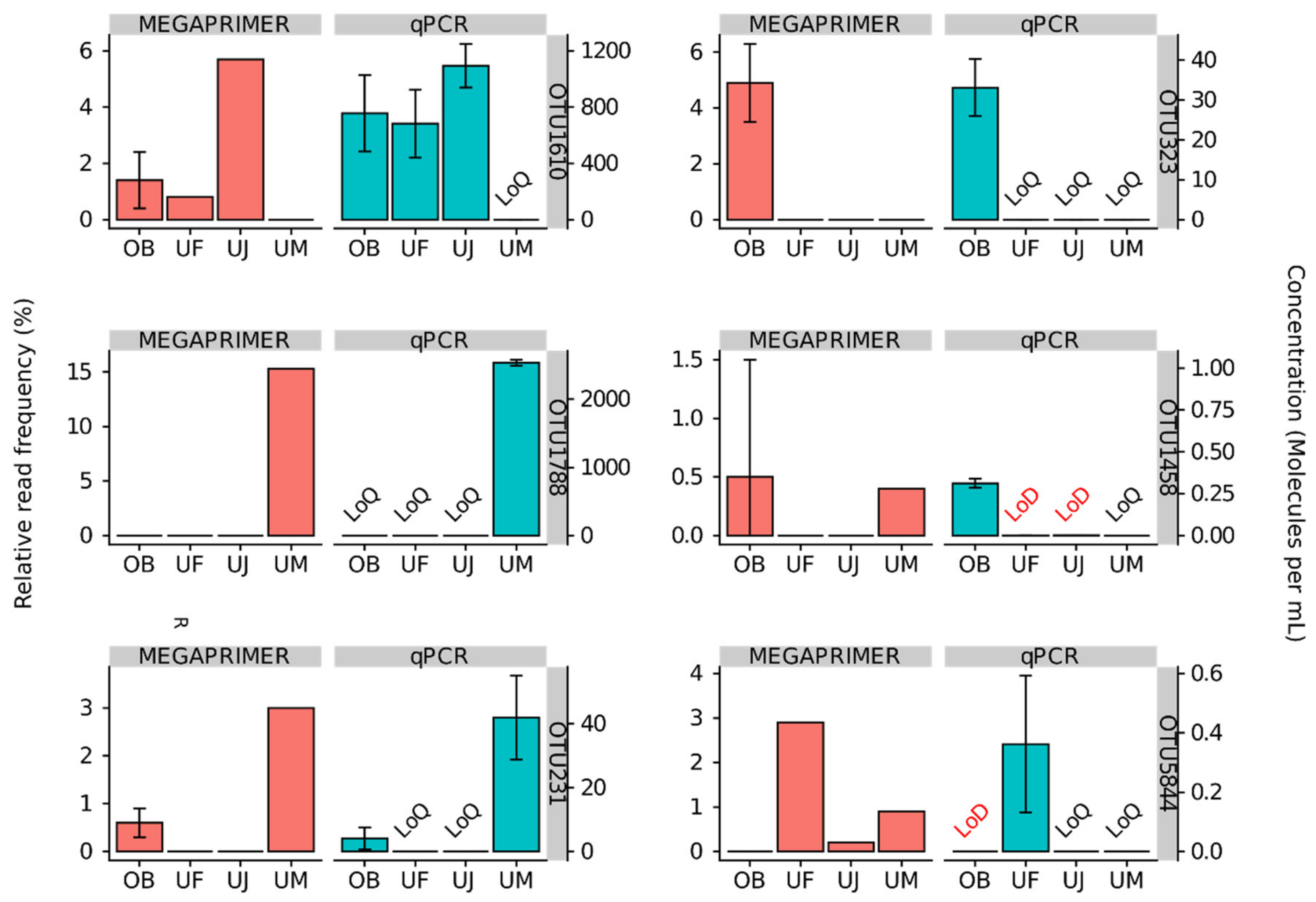
3.2. Quantitative Assessment of Mimiviridae Operational Taxonomic Units (OTUs) and its Comparison with Meta-Barcoding Profiles
3.3. Eukaryotic Communities
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Colson, P.; De Lamballerie, X.; Yutin, N.; Asgari, S.; Bigot, Y.; Bideshi, D.K.; Cheng, X.-W.; Federici, B.A.; Van Etten, J.L.; Koonin, E.V.; et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 2013, 158, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Scola, B.L.; Audic, S.; Robert, C.; Jungang, L.; de Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.-M.; Raoult, D. A Giant Virus in Amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Santini, S.; Rico, A.; Abergel, C.; Claverie, J.-M. Breaking the 1000-gene barrier for Mimivirus using ultra-deep genome and transcriptome sequencing. Virol. J. 2011, 8, 99. [Google Scholar] [CrossRef]
- Gallot-Lavallée, L.; Blanc, G.; Claverie, J.-M. Comparative Genomics of Chrysochromulina Ericina Virus and Other Microalga-Infecting Large DNA Viruses Highlights Their Intricate Evolutionary Relationship with the Established Mimiviridae Family. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Schvarcz, C.R.; Steward, G.F. A giant virus infecting green algae encodes key fermentation genes. Virology 2018, 518, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Needham, D.M.; Yoshizawa, S.; Hosaka, T.; Poirier, C.; Choi, C.J.; Hehenberger, E.; Irwin, N.A.T.; Wilken, S.; Yung, C.-M.; Bachy, C.; et al. A distinct lineage of giant viruses brings a rhodopsin photosystem to unicellular marine predators. Proc. Natl. Acad. Sci. 2019, 201907517. [Google Scholar] [CrossRef]
- Johannessen, T.V.; Bratbak, G.; Larsen, A.; Ogata, H.; Egge, E.S.; Edvardsen, B.; Eikrem, W.; Sandaa, R.-A. Characterisation of three novel giant viruses reveals huge diversity among viruses infecting Prymnesiales (Haptophyta). Virology 2015, 476, 180–188. [Google Scholar] [CrossRef]
- Abrahão, J.; Silva, L.; Silva, L.S.; Khalil, J.Y.B.; Rodrigues, R.; Arantes, T.; Assis, F.; Boratto, P.; Andrade, M.; Kroon, E.G.; et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 2018, 9, 749. [Google Scholar] [CrossRef]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. 2010, 107, 19508–19513. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Gann, E.R.; LeCleir, G.R.; Kang, Y.; Gobler, C.J.; Wilhelm, S.W. Diversity and dynamics of algal Megaviridae members during a harmful brown tide caused by the pelagophyte, Aureococcus anophagefferens. FEMS Microbiol. Ecol. 2016, 92, fiw058. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; LeCleir, G.R.; Brown, C.M.; Gobler, C.J.; Bidle, K.D.; Wilson, W.H.; Wilhelm, S.W. Genome of brown tide virus (AaV), the little giant of the Megaviridae, elucidates NCLDV genome expansion and host-virus coevolution. Virology 2014, 466–467, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, B.A.; Vladu, I.C.; Barclay, J.E.; Schroeder, D.C.; Malin, G.; Field, R.A. Isolation and Characterization of a Double Stranded DNA Megavirus Infecting the Toxin-Producing Haptophyte Prymnesium parvum. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Gallot-Lavallée, L.; Pagarete, A.; Legendre, M.; Santini, S.; Sandaa, R.-A.; Himmelbauer, H.; Ogata, H.; Bratbak, G.; Claverie, J.-M. The 474-Kilobase-Pair Complete Genome Sequence of CeV-01B, a Virus Infecting Haptolina (Chrysochromulina) ericina (Prymnesiophyceae). Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.J.; Nielsen, T.G.; Kaas, H. Distribution and growth of protists and mesozooplankton during a bloom of Chrysochromulina spp. (Prymnesiophyceae, Prymnesiales). Phycologia 1995, 34, 409–416. [Google Scholar] [CrossRef]
- Sandaa, R.-A.; Heldal, M.; Castberg, T.; Thyrhaug, R.; Bratbak, G. Isolation and Characterization of Two Viruses with Large Genome Size Infecting Chrysochromulina ericina (Prymnesiophyceae) and Pyramimonas orientalis (Prasinophyceae). Virology 2001, 290, 272–280. [Google Scholar] [CrossRef]
- Hingamp, P.; Grimsley, N.; Acinas, S.G.; Clerissi, C.; Subirana, L.; Poulain, J.; Ferrera, I.; Sarmento, H.; Villar, E.; Lima-Mendez, G.; et al. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 2013, 7, 1678–1695. [Google Scholar] [CrossRef]
- Mihara, T.; Koyano, H.; Hingamp, P.; Grimsley, N.; Goto, S.; Ogata, H. Taxon Richness of “Megaviridae” Exceeds those of Bacteria and Archaea in the Ocean. Microbes Environ. 2018, 33, 162–171. [Google Scholar] [CrossRef]
- Gran-Stadniczeñko, S.; Krabberød, A.K.; Sandaa, R.-A.; Yau, S.; Egge, E.; Edvardsen, B. Seasonal Dynamics of Algae-Infecting Viruses and Their Inferred Interactions with Protists. Viruses 2019, 11, 1043. [Google Scholar] [CrossRef]
- Chen, F.; Suttle, C.A. Amplification of DNA polymerase gene fragments from viruses infecting microalgae. Appl. Environ. Microbiol. 1995, 61, 1274–1278. [Google Scholar] [CrossRef]
- Chen, F.; Suttle, C.A.; Short, S.M. Genetic diversity in marine algal virus communities as revealed by sequence analysis of DNA polymerase genes. Appl. Environ. Microbiol. 1996, 62, 2869–2874. [Google Scholar] [CrossRef]
- Wilson, W.H.; Gilg, I.C.; Duarte, A.; Ogata, H. Development of DNA mismatch repair gene, MutS, as a diagnostic marker for detection and phylogenetic analysis of algal Megaviruses. Virology 2014, 466–467, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Endo, H.; Gotoh, Y.; Watai, H.; Ogawa, N.; Blanc-Mathieu, R.; Yoshida, T.; Ogata, H. The Earth Is Small for “Leviathans”: Long Distance Dispersal of Giant Viruses across Aquatic Environments. Microbes Environ. 2019, 34, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hingamp, P.; Watai, H.; Endo, H.; Yoshida, T.; Ogata, H.; Li, Y.; Hingamp, P.; Watai, H.; Endo, H.; et al. Degenerate PCR Primers to Reveal the Diversity of Giant Viruses in Coastal Waters. Viruses 2018, 10, 496. [Google Scholar] [CrossRef] [PubMed]
- Frias-Lopez, J.; Shi, Y.; Tyson, G.W.; Coleman, M.L.; Schuster, S.C.; Chisholm, S.W.; Delong, E.F. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. USA 2008, 105, 3805–3810. [Google Scholar] [CrossRef]
- Yoshida, T.; Yuki, Y.; Lei, S.; Chinen, H.; Yoshida, M.; Kondo, R.; Hiroishi, S. Quantitative Detection of Toxic Strains of the Cyanobacterial Genus Microcystis by Competitive PCR. Microbes Env. 2003, 18, 16–23. [Google Scholar] [CrossRef]
- Support Center. Available online: https://jp.support.illumina.com/?langsel=/jp/ (accessed on 17 February 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinforma. Oxf. Engl. 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinforma. Oxf. Engl. 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinforma. Oxf. Engl. 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Matsen, F.A.; Kodner, R.B.; Armbrust, E.V. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 2010, 11, 538. [Google Scholar] [CrossRef] [PubMed]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 5 February 2020).
- ggplot2 - Elegant Graphics for Data Analysis | Hadley Wickham | Springer. Available online: https://www.springer.com/us/book/9780387981413 (accessed on 23 February 2018).
- Hsieh, T.C.; Ma, K.H.; Chao, A. iNEXT: An R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PloS One 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ’omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Sólymos, P.; Stevens, M.H.H.; Wagner, H.; et al. vegan: Community Ecology Package. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 18 February 2020).
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinforma. Oxf. Engl. 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Forootan, A.; Sjöback, R.; Björkman, J.; Sjögreen, B.; Linz, L.; Kubista, M. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol. Detect. Quantif. 2017, 12, 1–6. [Google Scholar] [CrossRef]
- Bradley, I.M.; Pinto, A.J.; Guest, J.S. Design and Evaluation of Illumina MiSeq-Compatible, 18S rRNA Gene-Specific Primers for Improved Characterization of Mixed Phototrophic Communities. Appl. Env. Microbiol. 2016, 82, 5878–5891. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Jeudy, S.; Bartoli, J.; Poirot, O.; Lescot, M.; Abergel, C.; Barbe, V.; Wommack, K.E.; Noordeloos, A.A.M.; Brussaard, C.P.D.; et al. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc. Natl. Acad. Sci. USA 2013, 110, 10800–10805. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Sequencing Run Number | Sampling Location | Sampling Date | Primer Cocktail | Protocol Number1 |
|---|---|---|---|---|---|
| D-OB-MP5-1 | 1 | OB | 2015.10.30 | MP5 | 1 |
| D-OB-MP10-1 | 2 | OB | 2015.10.30 | MP10.v1 | 1 |
| D-OB-MP20-1 | 3 | OB | 2015.10.30 | MP20 | 1 |
| D-OB-MP1-2 | 4 | OB | 2015.10.30 | MP1 (no mix) | 2 |
| D-OB-MP5-2 | 5 | OB | 2015.10.30 | MP5 | 1 |
| D-OB-MP10-2 | 6 | OB | 2015.10.30 | MP10.v1 | 1 |
| D-OB-MP20-2 | 7 | OB | 2015.10.30 | MP20 | 1 |
| S-OB-MP10-1 | 8 | OB | 2015.10.30 | MP10.v2 | 3 |
| S-OB-MP10-2 | 8 | OB | 2015.10.30 | MP10.v1 | 1 |
| S-OB-MP10-3 | 8 | OB | 2015.10.30 | MP10.v2 | 4 |
| S-OB-MP10-4 | 8 | OB | 2015.10.30 | MP10.v1 | 5 |
| S-UF-MP10 | 8 | UF | 2017.6.21 | MP10.v2 | 3 |
| S-UJ-MP1 | 8 | UJ | 2017.7.6 | MP1 (no mix) | 3 |
| S-UJ-MP10 | 8 | UJ | 2017.7.6 | MP10.v2 | 3 |
| S-UM-MP10 | 8 | UM | 2017.11.10 | MP10.v2 | 3 |
| Dataset | Number of Raw Reads | Mimiviridae Reads | Proportion of Mimiviridae Reads | Number of OTUs | Primercocktail | Protocol Number |
|---|---|---|---|---|---|---|
| D-OB-MP1-0 [23] | 16,677,495 | 8,432,837 | 51% | 5,595 | MP1 (58/82 primer pairs) | - |
| D-OB-MP5-1 | 5,078,212 | 992,088 | 20% | 3,018 | MP5 | 1 |
| D-OB-MP10-1 | 5,995,548 | 1,916,193 | 32% | 3,396 | MP10.v1 | 1 |
| D-OB-MP20-1 | 10,720,091 | 1,019,645 | 10% | 3,110 | MP20 | 1 |
| D-OB-MP1-2 | 2,205,016 | 497,356 | 23% | 2,608 | MP1 | 2 |
| D-OB-MP5-2 | 2,992,984 | 273,153 | 9% | 2,426 | MP5 | 1 |
| D-OB-MP10-2 | 4,521,841 | 340,129 | 8% | 2,912 | MP10.v1 | 1 |
| D-OB-MP20-2 | 4,752,035 | 452,365 | 10% | 2,755 | MP20 | 1 |
| S-OB-MP10-1 | 60,348 | 5,258 | 9% | 744 | MP10.v2 | 3 |
| S-OB-MP10-2 | 78,067 | 37,638 | 48% | 1,487 | MP10.v1 | 1 |
| S-OB-MP10-3 | 34,860 | 11,942 | 34% | 1,243 | MP10.v2 | 4 |
| S-OB-MP10-4 | 38,477 | 21,965 | 57% | 1,388 | MP10.v1 | 5 |
| S-UF-MP10 | 96,149 | 29,275 | 30% | 601 | MP10.v2 | 3 |
| S-UJ-MP1 | 67,990 | 19,151 | 28% | 470 | MP1 | 3 |
| S-UJ-MP10 | 68,168 | 31,276 | 46% | 539 | MP10.v2 | 3 |
| S-UM-MP10 | 82,516 | 18,911 | 23% | 595 | MP10.v2 | 3 |
| Sampling Location | Number of Raw Reads | Taxonomically Annotated Reads | Number of OTUs |
|---|---|---|---|
| OB | 67,028 | 44,727 | 439 |
| UF | 95,352 | 63,833 | 325 |
| UJ | 81,281 | 67,479 | 285 |
| UM | 80,237 | 54,845 | 528 |
| Total | 323,898 | 230,884 | 1,156 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prodinger, F.; Endo, H.; Gotoh, Y.; Li, Y.; Morimoto, D.; Omae, K.; Tominaga, K.; Blanc-Mathieu, R.; Takano, Y.; Hayashi, T.; et al. An Optimized Metabarcoding Method for Mimiviridae. Microorganisms 2020, 8, 506. https://doi.org/10.3390/microorganisms8040506
Prodinger F, Endo H, Gotoh Y, Li Y, Morimoto D, Omae K, Tominaga K, Blanc-Mathieu R, Takano Y, Hayashi T, et al. An Optimized Metabarcoding Method for Mimiviridae. Microorganisms. 2020; 8(4):506. https://doi.org/10.3390/microorganisms8040506
Chicago/Turabian StyleProdinger, Florian, Hisashi Endo, Yasuhiro Gotoh, Yanze Li, Daichi Morimoto, Kimiho Omae, Kento Tominaga, Romain Blanc-Mathieu, Yoshihito Takano, Tetsuya Hayashi, and et al. 2020. "An Optimized Metabarcoding Method for Mimiviridae" Microorganisms 8, no. 4: 506. https://doi.org/10.3390/microorganisms8040506
APA StyleProdinger, F., Endo, H., Gotoh, Y., Li, Y., Morimoto, D., Omae, K., Tominaga, K., Blanc-Mathieu, R., Takano, Y., Hayashi, T., Nagasaki, K., Yoshida, T., & Ogata, H. (2020). An Optimized Metabarcoding Method for Mimiviridae. Microorganisms, 8(4), 506. https://doi.org/10.3390/microorganisms8040506