Beyond Just Bacteria: Functional Biomes in the Gut Ecosystem Including Virome, Mycobiome, Archaeome and Helminths

 , ,
, ,  , and
, and
Abstract
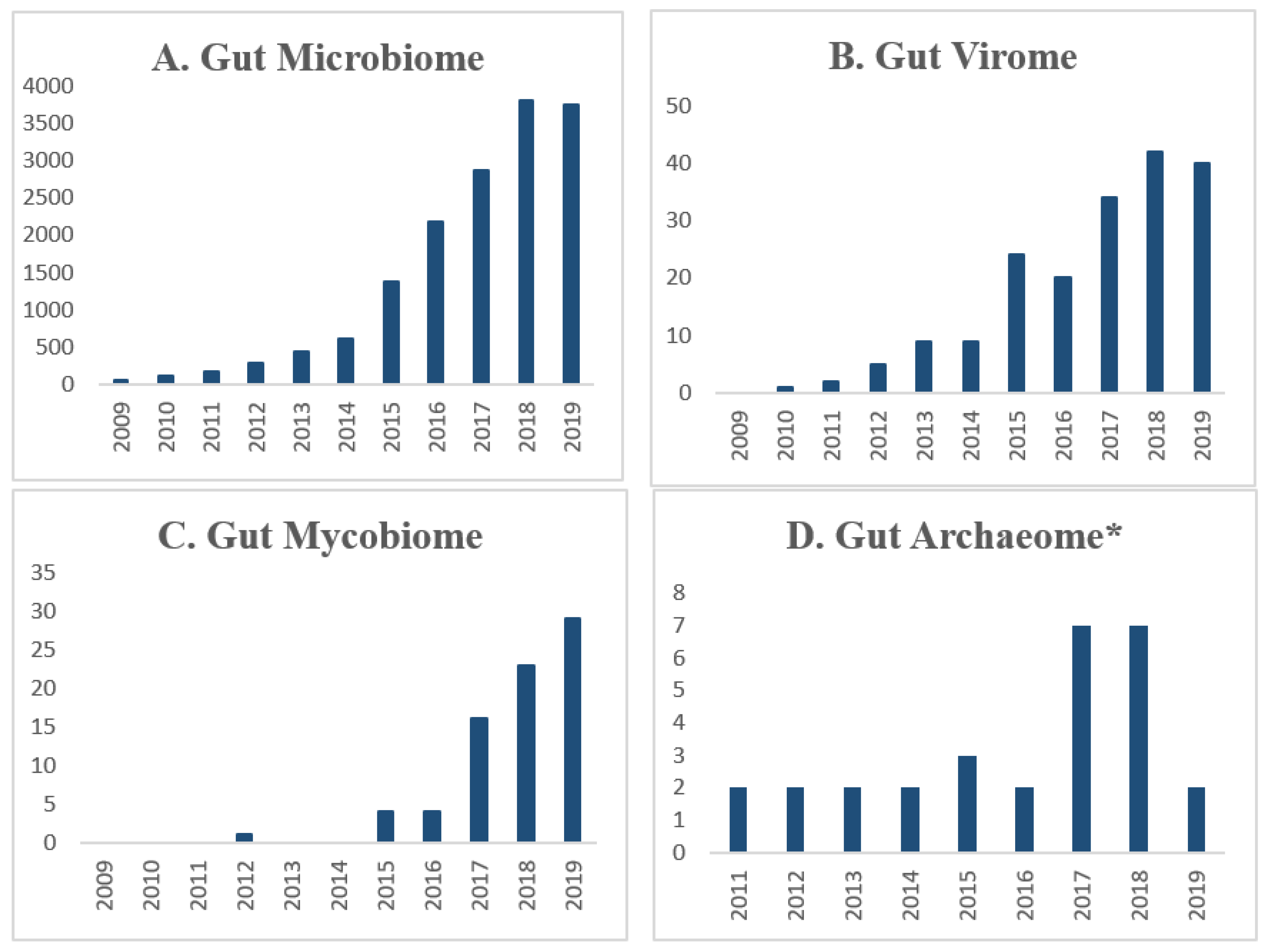
1. Introduction
2. Gut Virome
2.1. Gut Phages
2.2. Enteric Viruses
2.3. Role of the Gut Virome in Gastrointestinal Health and Disease
2.4. Viromes in Fecal Microbial Transplants and Fecal Virome Transplants
3. Mycobiome
4. Gut Archaea
5. Helminths
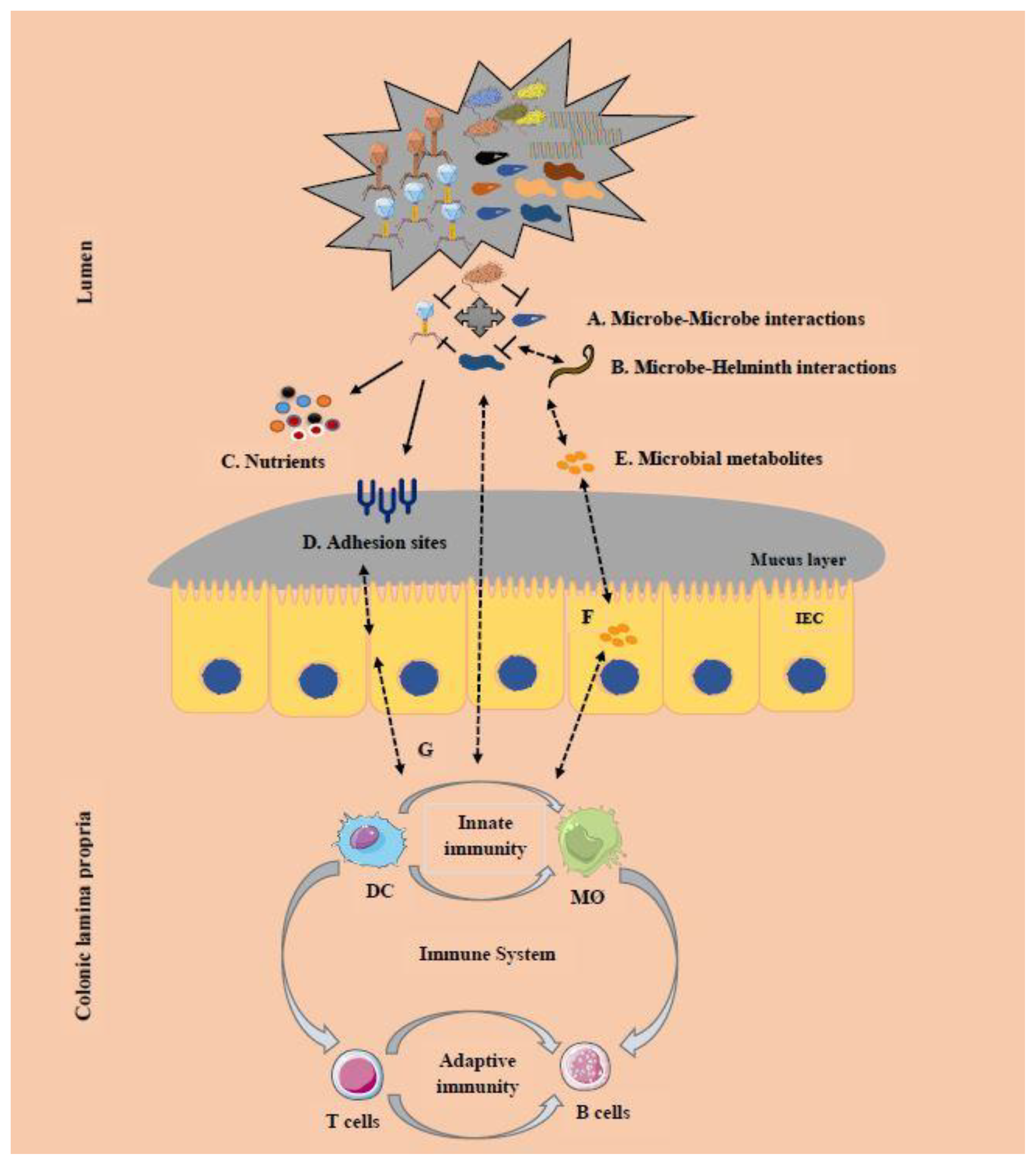
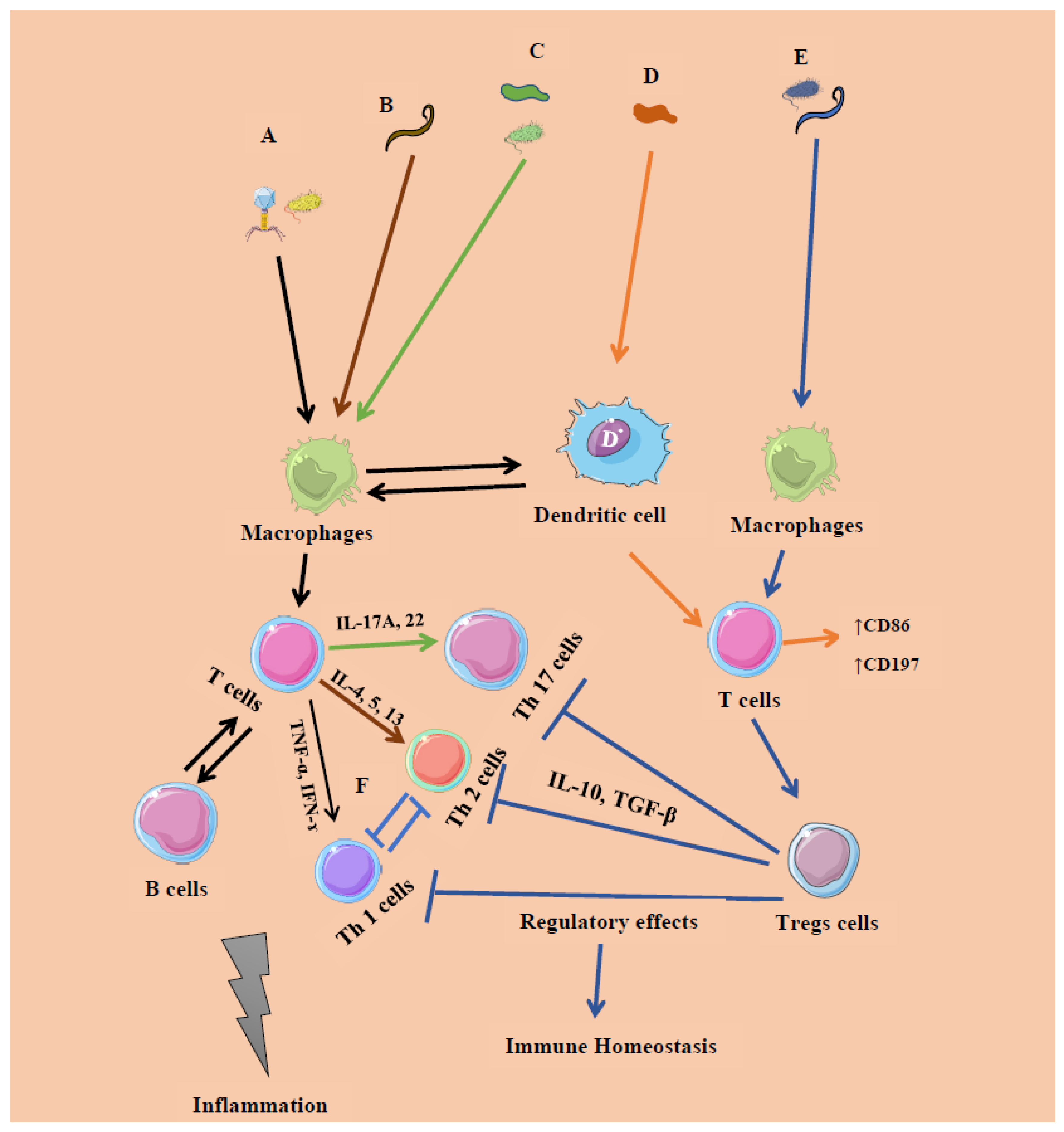
6. Cross-Kingdom Interactions
7. Modulation of the Microbiome and Related Concerns
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vemuri, R.; Gundamaraju, R.; Shastri, M.D.; Shukla, S.D.; Kalpurath, K.; Ball, M.; Tristram, S.; Shankar, E.M.; Ahuja, K.D.; Eri, R. Gut Microbial Changes, Interactions, and Their Implications on Human Lifecycle: An Ageing Perspective. BioMed Res. Int. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, R.; Gundamaraju, R.; Eri, R. Role of lactic acid probiotic bacteria in IBD. Curr. Pharm. Des. 2017, 23, 1. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, R.; Sylvia, K.; Klein, S.L.; Forster, S.C.; Plebanski, M.; Eri, R.; Flanagan, K.L. The microgenderome revealed: Sex differences in bidirectional interactions between the microbiota, hormones, immunity and disease susceptibility. Semin. Immunopathol. 2018, 41, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Kamm, M.A.; Colombel, J.-F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Seifert, M.; Pennhag, A.A.L.; Du, J.; Hamsten, M.C.; Schuppe-Koistinen, I.; Engstrand, L. A comprehensive automated pipeline for human microbiome sampling, 16S rRNA gene sequencing and bioinformatics processing. BioRxiv 2018, 286526. [Google Scholar] [CrossRef]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; González, A.; Kosciolek, T.; McCall, L.-I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Genet. 2018, 16, 410–422. [Google Scholar] [CrossRef]
- Stefanaki, C.; Peppa, M.; Mastorakos, G.; Chrousos, G.P. Examining the gut bacteriome, virome, and mycobiome in glucose metabolism disorders: Are we on the right track? Metabolism 2017, 73, 52–66. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Segal, J.P.; Carding, S.R.; Hart, A.L.; Hold, G.L. The gut virome: The ‘missing link’ between gut bacteria and host immunity? Ther. Adv. Gastroenterol. 2019, 12. [Google Scholar] [CrossRef]
- Forbes, J.D.; Bernstein, C.N.; Tremlett, H.; van Domselaar, G.; Knox, N. A Fungal World: Could the Gut Mycobiome Be Involved in Neurological Disease? Front. Microbiol. 2019, 9, 3249. [Google Scholar] [CrossRef]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2017, 3, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Witherden, E.; Moyes, D.L. Mycobiome and Gut Inflammation. In Immunity and Inflammation in Health and Disease; Elsevier: Amsterdam, The Netherlands, 2018; pp. 271–280. [Google Scholar]
- Gaci, N.; Borrel, G.; Tottey, W.; O’Toole, P.W.; Brugère, J.-F. Archaea and the human gut: New beginning of an old story. World J. Gastroenterol. 2014, 20, 16062–16078. [Google Scholar] [CrossRef] [PubMed]
- Giacomin, P.; Agha, Z.; Loukas, A. Helminths and Intestinal Flora Team Up to Improve Gut Health. Trends Parasitol. 2016, 32, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, J.V.; Elliott, D. Helminths and the IBD hygiene hypothesis. Inflamm. Bowel Dis. 2009, 15, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gallego, J.L.; Chou, S.-P.; Di Rienzi, S.C.; Goodrich, J.K.; Spector, T.D.; Bell, J.T.; Youngblut, N.D.; Hewson, I.; Reyes, A.; Ley, R.E. Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins. Cell Host Microbe 2019, 25, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.; Clooney, A.G.; Sutton, T.D.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.; Wong, M.C.; Smith, D.; Gesell, J.; Ross, M.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Motooka, D.; Fujimoto, K.; Tanaka, R.; Yaguchi, T.; Gotoh, K.; Maeda, Y.; Furuta, Y.; Kurakawa, T.; Goto, N.; Yasunaga, T.; et al. Fungal ITS1 Deep-Sequencing Strategies to Reconstruct the Composition of a 26-Species Community and Evaluation of the Gut Mycobiota of Healthy Japanese Individuals. Front. Microbiol. 2017, 8, 238. [Google Scholar] [CrossRef]
- Bhute, S.; Suryavanshi, M.V.; Joshi, S.M.; Yajnik, C.S.; Shouche, Y.S.; Ghaskadbi, S.S. Gut Microbial Diversity Assessment of Indian Type-2-Diabetics Reveals Alterations in Eubacteria, Archaea, and Eukaryotes. Front. Microbiol. 2017, 8, 399. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Raymann, K.; Moeller, A.H.; Goodman, A.L.; Ochman, H. Unexplored Archaeal Diversity in the Great Ape Gut Microbiome. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Chiu, C. Metagenomic Next-Generation Sequencing for Pathogen Detection and Identification. In Advanced Techniques in Diagnostic Microbiology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2018; pp. 617–632. [Google Scholar]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinform. 2014, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J. VIROME: A standard operating procedure for analysis of viral metagenome sequences. Stand. Genom. Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Soverini, M.; Turroni, S.; Quercia, S.; Biagi, E.; Brigidi, P.; Candela, M. ViromeScan: A new tool for metagenomic viral community profiling. BMC Genom. 2016, 17, 165. [Google Scholar] [CrossRef]
- Angly, F.; Rodriguez-Brito, B.; Bangor, D.; McNairnie, P.; Breitbart, M.; Salamon, P.; Felts, B.; Nulton, J.; Mahaffy, J.; Rohwer, F. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinform. 2005, 6, 41. [Google Scholar] [CrossRef]
- Leplae, R.; Lima-Mendez, G.; Toussaint, A. ACLAME: A CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2009, 38, D57–D61. [Google Scholar] [CrossRef]
- Zhao, G.; Wu, G.; Lim, E.S.; Droit, L.; Krishnamurthy, S.R.; Barouch, D.H.; Virgin, H.W.; Wang, D. VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology 2017, 503, 21–30. [Google Scholar] [CrossRef]
- Pal, R.R.; More, R.P.; Purohit, H.J. Bioinformatics Tools for Shotgun Metagenomic Data Analysis. In Soft Computing for Biological Systems; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2018; pp. 91–110. [Google Scholar]
- Barrientos-Somarribas, M.; Messina, D.; Pou, C.; Lysholm, F.; Bjerkner, A.; Allander, T.; Andersson, B.; Sonnhammer, E. Discovering viral genomes in human metagenomic data by predicting unknown protein families. Sci. Rep. 2018, 8, 28. [Google Scholar] [CrossRef]
- Du Toit, A. Nature Reviews Microbiology. Nat. Rev. Genet. 2019. [Google Scholar] [CrossRef]
- Shkoporov, A.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Brocal, V.P. The Human Virome; Springer: New York, NY, USA, 2018. [Google Scholar]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef] [PubMed]
- Abeles, S.R.; Pride, D.T. Molecular Bases and Role of Viruses in the Human Microbiome. J. Mol. Boil. 2014, 426, 3892–3906. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Park, E.-J.; Roh, S.W.; Bae, J.-W. Diversity and Abundance of Single-Stranded DNA Viruses in Human Feces. Appl. Environ. Microbiol. 2011, 77, 8062–8070. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gallego, J.L.; Chou, S.-P.; Di Rienzi, S.; Goodrich, J.K.; Spector, T.; Bell, J.T.; Youngblut, N.; Hewson, I.; Reyes, A.; Ley, R.E. The virome in adult monozygotic twins with concordant or discordant gut microbiomes. bioRxiv 2019. [Google Scholar] [CrossRef]
- Breitbart, M.; Haynes, M.; Kelley, S.; Angly, F.; Edwards, R.A.; Felts, B.; Mahaffy, J.M.; Mueller, J.; Nulton, J.; Rayhawk, S.; et al. Viral diversity and dynamics in an infant gut. Res Microbiol 2008, 159, 367–373. [Google Scholar] [CrossRef]
- Kolde, R.; Franzosa, E.A.; Rahnavard, G.; Hall, A.B.; Vlamakis, H.; Stevens, C.; Daly, M.J.; Xavier, R.J.; Huttenhower, C. Host genetic variation and its microbiome interactions within the Human Microbiome Project. Genome Med. 2018, 10, 6. [Google Scholar] [CrossRef]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef]
- Reyes, A.; Blanton, L.V.; Cao, S.; Zhao, G.; Manary, M.; Trehan, I.; Smith, M.I.; Wang, D.; Virgin, H.W.; Rohwer, F.; et al. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA 2015, 112, 11941–11946. [Google Scholar] [CrossRef] [PubMed]
- Maya-Lucas, O.; Murugesan, S.; Nirmalkar, K.; Alcaraz, L.D.; Hoyo-Vadillo, C.; Pizano-Zárate, M.L.; García-Mena, J.J.A. The gut microbiome of Mexican children affected by obesity. Anaerobe 2019, 55, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Breitbart, M.; Lee, W.H.; Run, J.-Q.; Wei, C.L.; Soh, S.W.L.; Hibberd, M.L.; Liu, E.T.; Rohwer, F.; Ruan, Y. RNA viral community in human feces: Prevalence of plant pathogenic viruses. PLoS Biol. 2005, 4, e3. [Google Scholar] [CrossRef]
- Stockdale, S.R.; Ryan, F.J.; McCann, A.; Dalmasso, M.; Ross, P.R.; Hill, C. Viral Dark Matter in the Gut Virome of Elderly Humans. Preprints 2018. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.J.C. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Ma, Y.; You, X.; Mai, G.; Tokuyasu, T.; Liu, C.J.M. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome 2018, 6, 24. [Google Scholar] [CrossRef]
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.J.; et al. Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe 2016, 19, 311–322. [Google Scholar] [CrossRef]
- D’arc, M.; Furtado, C.; Siqueira, J.D.; Seuánez, H.N.; Ayouba, A.; Peeters, M.; Soares, M.A. Assessment of the gorilla gut virome in association with natural simian immunodeficiency virus infection. Retrovirology 2018, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Yu, H.; Xu, L.; Zhao, Z.; Zhang, P.; Qu, Y.; He, B.; Tu, C. Virome profiling of rodents in Xinjiang Uygur Autonomous Region, China: Isolation and characterization of a new strain of Wenzhou virus. Virology 2019, 529, 122–134. [Google Scholar] [CrossRef]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog. 2011, 7, e1002218. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.A.; Mollerup, S.; Nguyen, N.P.; White, N.E.; Coghlan, M.; Alquezar-Planas, D.E.; Joshi, T.; Jensen, R.H.; Fridholm, H.; Kjartansdóttir, K.R.; et al. High diversity of picornaviruses in rats from different continents revealed by deep sequencing. Emerging Microbes Infections 2016, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.J.M. Viral diversity of house mice in New York City. Am. Soc. Microbiol. 2018, 9, e01354-17. [Google Scholar] [CrossRef]
- Duerkop, B.A.; Kleiner, M.; Paez-Espino, D.; Zhu, W.; Bushnell, B.; Hassell, B.; Winter, S.E.; Kyrpides, N.C.; Hooper, L.V. Murine colitis reveals a disease-associated bacteriophage community. Nat. Microbil. 2018, 3, 1023. [Google Scholar] [CrossRef]
- Seth, R.; Maqsood, R.; Mondal, A.; Bose, D.; Kimono, D.; Holland, L.; Janulewicz, P.A.; Klimas, N.; Horner, R.D.; Sullivan, K.; et al. Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness. Viruses 2019, 11, 968. [Google Scholar] [CrossRef]
- Reyes, A.; Wu, M.; McNulty, N.P.; Rohwer, F.L.; Gordon, J.I. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. USA 2013, 110, 20236–20241. [Google Scholar] [CrossRef]
- Minot, S.S.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Conservation of Gene Cassettes among Diverse Viruses of the Human Gut. PLoS ONE 2012, 7, e42342. [Google Scholar] [CrossRef]
- Finkbeiner, S.R.; Allred, A.F.; Tarr, P.I.; Klein, E.J.; Kirkwood, C.D.; Wang, D. Metagenomic Analysis of Human Diarrhea: Viral Detection and Discovery. PLOS Pathog. 2008, 4, e1000011. [Google Scholar] [CrossRef]
- Ogilvie, L.; Jones, B.V. The human gut virome: A multifaceted majority. Front. Microbiol. 2015, 6, 918. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Ianiro, G.; Scaldaferri, F.; Cammarota, G.; Gasbarrini, A. Gut Virome and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 1708–1712. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Härkönen, T.; Hämäläinen, A.-M.; et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [PubMed]
- Waller, A.S.; Yamada, T.; Kristensen, D.M.; Kultima, J.R.; Sunagawa, S.; Koonin, E.V.; Bork, P. Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J. 2014, 8, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Da Costa, A.C.; Del Valle-Mendoza, J.; Bucardo-Rivera, F.; Nordgren, J.; O’Ryan, M.; Deng, X.; Delwart, E. The fecal virome of South and Central American children with diarrhea includes small circular DNA viral genomes of unknown origin. Arch. Virol. 2016, 161, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.L.; Schapendonk, C.M.; Van Beek, J.; Vennema, H.; Schürch, A.C.; Schipper, D.; Bodewes, R.; Haagmans, B.L.; Osterhaus, A.D.M.E.; Koopmans, M. New Viruses in Idiopathic Human Diarrhea Cases, the Netherlands. Emerg. Infect. Dis. 2014, 20, 1218–1222. [Google Scholar] [CrossRef]
- Park, A.; Zhao, G. Mining the Virome for Insights into Type 1 Diabetes. DNA Cell Boil. 2018, 37, 422–425. [Google Scholar] [CrossRef]
- Chehoud, C.; Dryga, A.; Hwang, Y.; Nagy-Szakal, R.; Hollister, E.B.; Luna, R.A.; Versalovic, J.; Kellermayer, R.; Bushman, F.D. Transfer of Viral Communities between Human Individuals during Fecal Microbiota Transplantation. mBio 2016, 7, e00322-16. [Google Scholar] [CrossRef]
- Zuo, T.; Wong, S.H.; Lam, K.; Lui, R.; Cheung, K.; Tang, W.; Ching, J.Y.L.; Chan, P.K.S.; Chan, M.; Wu, J.C.Y.; et al. Bacteriophage transfer during faecal microbiota transplantation inClostridium difficileinfection is associated with treatment outcome. Gut 2017, 67, 634–643. [Google Scholar] [CrossRef]
- Draper, L.A.; Ryan, F.; Smith, M.K.; Jalanka, J.; Mattila, E.; Arkkila, P.A.; Ross, R.; Satokari, R.; Hill, C. Long-term colonisation with donor bacteriophages following successful faecal microbial transplantation. Microbiome 2018, 6, 220. [Google Scholar] [CrossRef]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Anderson, J.M.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients With Clostridium difficile Infection. Gastroenterol. 2017, 152, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.-D.; Pang, M.-D.; Olaniran, A.; Zhang, X.-H.; Zhang, H.; Zhou, Y.; Sun, L.-C.; Schmidt, S.; Wang, R. Alterations in the diversity and composition of mice gut microbiota by lytic or temperate gut phage treatment. Appl. Microbiol. Biotechnol. 2018, 102, 10219–10230. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.M.; Koskella, B.; Ritz, N.L.; Lin, D.; Carroll-Portillo, A.; Lin, H.C. Transplanting Fecal Virus-Like Particles Reduces High-Fat Diet-Induced Small Intestinal Bacterial Overgrowth in Mice. Front. Microbiol. 2019, 9, 348. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.S.; Mentzel, C.M.J.; Kot, W.; Castro-Mejía, J.L.; Zuffa, S.; Swann, J.R.; Hansen, L.H.; Vogensen, F.K.; Hansen, A.K.; Nielsen, D.S. Faecal virome transplantation decreases symptoms of type 2 diabetes and obesity in a murine model. Gut 2020. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Hsu, B.B.; Gibson, T.E.; Yeliseyev, V.; Liu, Q.; Lyon, L.; Bry, L.; Silver, P.A.; Gerber, G.K. Dynamic Modulation of the Gut Microbiota and Metabolome by Bacteriophages in a Mouse Model. Cell Host Microbe 2019, 25, 803–814. [Google Scholar] [CrossRef]
- Conceição-Neto, N.; Deboutte, W.; Dierckx, T.; Machiels, K.; Wang, J.; Yinda, K.C.; Maes, P.; Van Ranst, M.; Joossens, M.; Raes, J.; et al. Low eukaryotic viral richness is associated with faecal microbiota transplantation success in patients with UC. Gut 2017, 67, 1558–1559. [Google Scholar] [CrossRef]
- Jones, L.; Kumar, J.; Mistry, A.; Sankar Chittoor Mana, T.; Perry, G.; Reddy, V.P.; Obrenovich, M. The Transformative Possibilities of the Microbiota and Mycobiota for Health, Disease, Aging, and Technological Innovation. Biomedicines 2019, 7, 24. [Google Scholar] [CrossRef]
- Tang, J.; Iliev, I.D.; Brown, J.; Underhill, D.M.; Funari, V.A. Mycobiome: Approaches to Analysis of Intestinal Fungi. J. Immunol. Methods 2015, 421, 112–121. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.-H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi - recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; NIH Intramural Sequencing Center Comparative Sequencing Program; et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, M.; Fosso, B.; Consiglio, A.; De Caro, G.; Grillo, G.; Licciulli, F.; Liuni, S.; Marzano, M.; Alonso-Alemany, D.; Valiente, G.; et al. Reference databases for taxonomic assignment in metagenomics. Briefings Bioinform. 2012, 13, 682–695. [Google Scholar] [CrossRef]
- Koetschan, C.; Hackl, T.; Müller, T.; Wolf, M.; Förster, F.; Schultz, J. ITS2 Database IV: Interactive taxon sampling for internal transcribed spacer 2 based phylogenies. Mol. Phylogenetics Evol. 2012, 63, 585–588. [Google Scholar] [CrossRef]
- Schoch, C.L.; Robbertse, B.; Robert, V.; Vu, D.; Cardinali, G.; Irinyi, L.; Meyer, W.; Nilsson, R.H.; Hughes, K.; Miller, A.N.; et al. Finding needles in haystacks: Linking scientific names, reference specimens and molecular data for Fungi. Database 2014. [Google Scholar] [CrossRef]
- Irinyi, L.; Serena, C.; Garcia-Hermoso, D.; Arabatzis, M.; Desnos-Ollivier, M.; Vu, D.; Cardinali, G.; Arthur, I.; Normand, A.-C.; Giraldo, A.; et al. International Society of Human and Animal Mycology (ISHAM)-ITS reference DNA barcoding database—The quality controlled standard tool for routine identification of human and animal pathogenic fungi. Med Mycol. 2015, 53, 313–337. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Standaert, A.; Sendid, B.; Joossens, M.; François, N.; Khoury, P.V.-E.; Branche, J.; Van Kruiningen, H.; Jouault, T.; Rutgeerts, P.; Gower, C.; et al. Candida albicans Colonization and ASCA in Familial Crohn’s Disease. Am. J. Gastroenterol. 2009, 104, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol. Boil. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2016, 8, 352–358. [Google Scholar] [CrossRef]
- Wheeler, M.L.; Limon, J.J.; Bar, A.S.; Leal, C.A.; Gargus, M.; Tang, J.; Brown, J.; Funari, V.A.; Wang, H.L.; Crother, T.R.; et al. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe 2016, 19, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2016, 66, 1039–1048. [Google Scholar] [CrossRef]
- Limon, J.J.; Skalski, J.; Underhill, D.M. Commensal Fungi in Health and Disease. Cell Host Microbe 2017, 22, 156–165. [Google Scholar] [CrossRef]
- Lai, G.C.; Tan, T.G.; Pavelka, N. The mammalian mycobiome: A complex system in a dynamic relationship with the host. Wiley Interdiscip. Rev. Syst. Boil. Med. 2018, 11, e1438. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and Fungi of the Human Gut Microbiome: Correlations with Diet and Bacterial Residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [PubMed]
- Schei, K.; Avershina, E.; Øien, T.; Rudi, K.; Follestad, T.; Salamati, S.; Ødegård, R.A. Early gut mycobiota and mother-offspring transfer. Microbiome 2017, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Rosin, M.; Lee, A.; Boxen, K.; Duncan, A.; Rosendale, D.; Villas-Boas, S.; McKenzie, E.J. The New Zealand human gut mycobiome. In Proceedings of the Foodomics 2019 High Value Nutrition National Science Challenge, Auckland, New Zealand, 9–11 April 2019. [Google Scholar]
- Ahmad, H.F.B.; Mejia, J.L.C.; Kot, W.; Bechshøft, R.L.; Reitelseder, S.; Holm, L.; Nielsen, D.S. The Gut Mycobiome of Elderly Danes. In Proceedings of the Danish Microbiological Society Annual Congress 2016, Comenhagen, Denmark, 14 November 2016. [Google Scholar]
- Borges, F.M.; De Paula, T.O.; Sarmiento, M.R.A.; De Oliveira, M.G.; Pereira, M.L.M.; Toledo, I.V.; Nascimento, T.C.; Ferreira-Machado, A.B.; Silva, V.L.; Diniz, C.G. Fungal Diversity of Human Gut Microbiota Among Eutrophic, Overweight, and Obese Individuals Based on Aerobic Culture-Dependent Approach. Curr. Microbiol. 2018, 75, 726–735. [Google Scholar] [CrossRef]
- Botschuijver, S.; Roeselers, G.; Levin, E.; Jonkers, D.M.; Welting, O.; Heinsbroek, S.E.M.; De Weerd, H.H.; Boekhout, T.; Fornai, M.; Masclee, A.A.; et al. Intestinal Fungal Dysbiosis Is Associated With Visceral Hypersensitivity in Patients With Irritable Bowel Syndrome and Rats. Gastroenterol. 2017, 153, 1026–1039. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Jiang, H.; Yuan, L.; Zhang, L.; Ma, J.-E.; Zhang, X.; Cheng, M.; Chen, J. Fecal Bacteriome and Mycobiome in Bats with Diverse Diets in South China. Curr. Microbiol. 2018, 75, 1352–1361. [Google Scholar] [CrossRef]
- Foster, M.L.; Dowd, S.E.; Stephenson, C.; Steiner, J.M.; Suchodolski, J.S. Characterization of the Fungal Microbiome (Mycobiome) in Fecal Samples from Dogs. Veter. Med. Int. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Huffman, M.A.; Li, J.-H. The Gut Microbiome of Tibetan Macaques: Composition, Influencing Factors and Function in Feeding Ecology. In The Evolution of Feathers; Springer Science and Business Media LLC: Berlin/Heilderbeg, Germany, 2019; pp. 207–222. [Google Scholar]
- Lurie-Weinberger, M.N.; Gophna, U. Archaea in and on the Human Body: Health Implications and Future Directions. PLOS Pathog. 2015, 11, e1004833. [Google Scholar] [CrossRef] [PubMed]
- Dridi, B.; Fardeau, M.-L.; Ollivier, B.; Raoult, D.; Drancourt, M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2012, 62, 1902–1907. [Google Scholar] [CrossRef]
- Dridi, B.; Henry, M.; El Khéchine, A.; Raoult, D.; Drancourt, M. High Prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae Detected in the Human Gut Using an Improved DNA Detection Protocol. PLoS ONE 2009, 4, e7063. [Google Scholar] [CrossRef] [PubMed]
- Mihajlovski, A.; Doré, J.; Levenez, F.; Alric, M.; Brugère, J.-F. Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ. Microbiol. Rep. 2010, 2, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, R.; Shinde, T.; Shastri, M.D.; Perera, A.P.; Tristram, S.; Martoni, C.J.; Gundamaraju, R.; Ahuja, K.D.K.; Ball, M.; Eri, R. A human origin strain Lactobacillus acidophilus DDS-1 exhibits superior in vitro probiotic efficacy in comparison to plant or dairy origin probiotics. Int. J. Med Sci. 2018, 15, 840–848. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Pimentel, M.; Lin, H.; Enayati, P.; Burg, B.V.D.; Lee, H.-R.; Chen, J.H.; Park, S.; Kong, Y.; Conklin, J. Methane, a gas produced by enteric bacteria, slows intestinal transit and augments small intestinal contractile activity. Am. J. Physiol. Liver Physiol. 2006, 290, G1089–G1095. [Google Scholar] [CrossRef]
- Lurie-Weinberger, M.N.; Peeri, M.; Tuller, T.; Gophna, U. Extensive Inter-Domain Lateral Gene Transfer in the Evolution of the Human Commensal Methanosphaera stadtmanae. Front. Genet. 2012, 3, 182. [Google Scholar] [CrossRef]
- Lurie-Weinberger, M.N.; Peeri, M.; Gophna, U. Contribution of lateral gene transfer to the gene repertoire of a gut-adapted methanogen. Genome 2012, 99, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.; Weidenbach, K.; Gutsmann, T.; Heine, H.; Schmitz-Streit, R.A. The Intestinal Archaea Methanosphaera stadtmanae and Methanobrevibacter smithii Activate Human Dendritic Cells. PLoS ONE 2014, 9, e99411. [Google Scholar] [CrossRef] [PubMed]
- Oxley, A.P.A.; Lanfranconi, M.P.; Würdemann, D.; Ott, S.; Stefánsson, K.; McGenity, T.J.; Timmis, K.N.; Nogales, B. Halophilic archaea in the human intestinal mucosa. Environ. Microbiol. 2010, 12, 2398–2410. [Google Scholar] [CrossRef]
- Wampach, L.; Heintz-Buschart, A.; Hogan, A.; Muller, E.; Narayanasamy, S.; Laczny, C.C.; Hugerth, L.W.; Bindl, L.; Bottu, J.; Andersson, A.F.; et al. Colonization and Succession within the Human Gut Microbiome by Archaea, Bacteria, and Microeukaryotes during the First Year of Life. Front. Microbiol. 2017, 8, 434. [Google Scholar] [CrossRef] [PubMed]
- Mbakwa, C.A.; Penders, J.; Savelkoul, P.H.; Thijs, C.; Dagnelie, P.C.; Mommers, M.; Arts, I.C. Gut colonization with Methanobrevibacter smithii is associated with childhood weight development. Obesity 2015, 23, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Abell, G.C.; Conlon, M.A.; McOrist, A.L. Methanogenic archaea in adult human faecal samples are inversely related to butyrate concentration. Microb. Ecol. Heal. Dis. 2006, 18, 154–160. [Google Scholar]
- Jalanka, J.; Salonen, A.; Nikkilä, J.; Immonen, O.; Kekkonen, R.; Lahti, L.; Palva, A.; De Vos, W.M. Intestinal Microbiota in Healthy Adults: Temporal Analysis Reveals Individual and Common Core and Relation to Intestinal Symptoms. PLoS ONE 2011, 6, e23035. [Google Scholar]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.-J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Dridi, B.; Henry, M.; Richet, H.; Raoult, D.; Drancourt, M.J.A. Age-related prevalence of M ethanomassiliicoccus luminyensis in the human gut microbiome. Apmis 2012, 120, 773–777. [Google Scholar] [CrossRef]
- Borrel, G.; McCann, A.; Deane, J.; Neto, M.C.; Lynch, D.B.; Brugère, J.-F.; O’Toole, P.W. Genomics and metagenomics of trimethylamine-utilizing Archaea in the human gut microbiome. ISME J. 2017, 11, 2059–2074. [Google Scholar] [CrossRef]
- Lecours, P.B.; Marsolais, D.; Cormier, Y.; Berberi, M.; Haché, C.; Bourdages, R.; Duchaine, C. Increased Prevalence of Methanosphaera stadtmanae in Inflammatory Bowel Diseases. PLoS ONE 2014, 9, e87734. [Google Scholar] [CrossRef]
- Ghavami, S.B.; Rostami, E.; Sephay, A.A.; Shahrokh, S.; Balaii, H.; Aghdaei, H.A.; Zali, M.R. Alterations of the human gut Methanobrevibacter smithii as a biomarker for inflammatory bowel diseases. Microb. Pathog. 2018, 117, 285–289. [Google Scholar] [CrossRef]
- Zhu, Y.; Sun, Y.; Wang, C.; Li, F. Impact of dietary fibre: Starch ratio in shaping caecal archaea revealed in rabbits. J. Anim. Physiol. Anim. Nutr. 2016, 101, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, J.V.; Summers, R.; Elliott, D. Helminths and harmony. Gut 2004, 53, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Maizels, R.M. Diversity and dialogue in immunity to helminths. Nat. Rev. Immunol. 2011, 11, 375–388. [Google Scholar] [CrossRef]
- Weinstock, J.V.; Summers, R.W.; Elliott, D.; Qadir, K.; Urban, J.F.; Thompson, R. The possible link between de-worming and the emergence of immunological disease. J. Lab. Clin. Med. 2002, 139, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Giacomin, P.; Croese, J.; Krause, L.; Loukas, A.; Cantacessi, C. Suppression of inflammation by helminths: A role for the gut microbiota? Philos. Trans. R. Soc. B: Boil. Sci. 2015, 370, 20140296. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, D.; Bowcutt, R.; Lee, S.C.; Tang, M.S.; Kurtz, Z.D.; Ding, Y.; Honda, K.; Gause, W.C.; Blaser, M.J.; Bonneau, R.A.; et al. Helminth infection promotes colonization resistance via type 2 immunity. Science 2016, 352, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Helmby, H. Human helminth therapy to treat inflammatory disorders—Where do we stand? BMC Immunol. 2015, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Summers, R.W.; Elliott, D.; Urban, J.F.; Thompson, R.A.; Weinstock, J.V. Trichuris suis therapy for active ulcerative colitis: A randomized controlled trial. Gastroenterol. 2005, 128, 825–832. [Google Scholar] [CrossRef]
- Moreels, T.G.; Nieuwendijk, R.J.; De Man, J.G.; De Winter, B.Y.; Herman, A.G.; Van Marck, E.A.; Pelckmans, P.A. Concurrent infection with Schistosoma mansoni attenuates inflammation induced changes in colonic morphology, cytokine levels, and smooth muscle contractility of trinitrobenzene sulphonic acid induced colitis in rats. Gut 2004, 53, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, M.J.; Ardeshir, A.; Kanwar, B.; Mirpuri, J.; Gundra, U.M.; Leung, J.; Wiens, K.; Vujkovic-Cvijin, I.; Kim, C.C.; Yarovinsky, F.; et al. Therapeutic Helminth Infection of Macaques with Idiopathic Chronic Diarrhea Alters the Inflammatory Signature and Mucosal Microbiota of the Colon. PLOS Pathog. 2012, 8, e1003000. [Google Scholar] [CrossRef] [PubMed]
- Sabin, E.A.; Carvalho, E.M.; Araujo, M.I.; Pearce, E.J. Impairment of Tetanus Toxoid-Specific Thl-like Immune Responses in Humans Infected with Schistosoma mansoni. J. Infect. Dis. 1996, 173, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Summers, R.W.; Elliott, D.E.; Qadir, K.; Urban, J.F., Jr.; Thompson, R.; Weinstock, J.V. Trichuris suis seems to be safe and possibly effective in the treatment of inflammatory bowel disease. Am. J. Gastoeneorol. 2003, 98, 2034. [Google Scholar] [CrossRef]
- Sandborn, W.; Elliott, D.; Weinstock, J.; Summers, R.; Landry-Wheeler, A.; Silver, N.; Harnett, M.; Hanauer, S. Randomised clinical trial: The safety and tolerability of T richuris suis ova in patients with Crohn’s disease. Aliment Pharnacol. Ther. 2013, 38, 255–263. [Google Scholar] [CrossRef]
- Croese, J.; O’neil, J.; Masson, J.; Cooke, S.; Melrose, W.; Pritchard, D.; Speare, R. A proof of concept study establishing Necator americanus in Crohn’s patients and reservoir donors. Gut 2006, 55, 136–137. [Google Scholar] [CrossRef]
- Fleming, J.O.; Isaák, A.; Lee, J.E.; Luzzio, C.C.; Carrithers, M.D.; Cook, T.D.; Field, A.S.; Boland, J.; Fabry, Z. Probiotic helminth administration in relapsing—Remitting multiple sclerosis: A phase 1 study. Mult. Scler. J. 2011, 17, 743–754. [Google Scholar] [CrossRef]
- Ghoul, M.; Mitri, S. The Ecology and Evolution of Microbial Competition. Trends Microbiol. 2016, 24, 833–845. [Google Scholar] [CrossRef]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Genet. 2010, 8, 15–25. [Google Scholar] [CrossRef]
- Zuo, T.; Wong, S.H.; Cheung, C.P.; Lam, K.; Lui, R.; Cheung, K.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Wu, J.C.Y.; et al. Gut fungal dysbiosis correlates with reduced efficacy of fecal microbiota transplantation in Clostridium difficile infection. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Karst, S.M. The influence of commensal bacteria on infection with enteric viruses. Nat. Rev. Genet. 2016, 14, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Filyk, H.A.; Osborne, L.C. The Multibiome: The Intestinal Ecosystem’s Influence on Immune Homeostasis, Health, and Disease. EBioMedicine 2016, 13, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Lepaslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.D.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Ouwehand, A.C. A review of dose-responses of probiotics in human studies. Benef. Microbes 2017, 8, 143–151. [Google Scholar] [CrossRef]
- Joseph, J.; Law, C. Cross-species examination of single- and multi-strain probiotic treatment effects on neuropsychiatric outcomes. Neurosci. Biobehav. Rev. 2019, 99, 160–197. [Google Scholar] [CrossRef]
- Kearney, S.M.; Gibbons, S.M. Designing synbiotics for improved human health. Microb. Biotechnol. 2017, 11, 141–144. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Microbial Component | Samples | Sorting/Analysis Method | Reads/Contigs */Sequences | Ref. |
|---|---|---|---|---|
| Viral | 21 | Hidden Markov Models/STAR | Assembled: 107,307 contigs. Total taxonomically assigned: 12,751 contigs (29.62% only) | [17] |
| 10 | Assembled: 294,211 contigs Alignment of reads to contigs: 57,721 Final catalogue: 39,254 contigs | [18] | ||
| 32 | CD-Hit-est | Total reads: 1,386,331 (32 datasets) Date normalized: 14,000 reads/dataset Annotated yield: 5004 contigs | [19] | |
| Fungal | 317 (147 subjects) | Internal Transcribed Spacer 2 (ITS) | Total reads per 1 sample/subject: 756,316 reads Total reads/sample: 17,189 | [20] |
| 14 | ITS1 (IonPGM, MiSeq, PacBio Sequence comparisons) | IonPGM: 219,756 reads MiSeq: 181,436 reads PacBio: 2984/sample Total: 41,776 reads | [21] | |
| 49 | ITS | 106,185 reads | [22] | |
| Archaeal (Methanogens) | 21 | 16S gene analysis | 1521 sequences | [23] |
| 10 | 10,000 reads/sample | [24] | ||
| 49 | 109,561 reads | [22] |
| Model Type | Study Type | Findings | Ref |
|---|---|---|---|
| Human | |||
| Healthy infant | 1 week old infant N = 1 | Taxa identified: Siphoviruses and prophages (the majority (72%) | [41] |
| Infants/mothers (healthy) | Healthy adult female monozygotic co-twins and their mothers at three time points over a one-year period N = 12 | Eukaryotic viral genomes: 73.3%, phages and prophages: 25.8% dsDNA phage (Caudovirales): 76.9% | [19] |
| N = 8 (4 twin pairs) | Taxa identified: Siphoviridae, Inoviridae, Myoviridae and Podoviridae | [43] | |
| N = 24 longitudinal fecal samples | Taxa identified: Microviridae, Podoviridae, Myoviridae, and Siphoviridae | [44] | |
| Healthy vs. malnourished infants/children | Time-series from fecal samples (Malawian) healthy control (HC)= twins, Mal Nutr = 12 twin pairs | ↑ Anelloviridae (ssDNA eukaryotic viruses) in healthy infants and children (up to 15–18 mo), With age: ↑ Alpavirinae (ssDNA phages), ↑ Siphoviridae in 0 to 10 mo of age and then slowly decrease. | [45] |
| Obese children | N = 20 (HC = 10, Obese = 10) | Obese to HC: ↑ Human herpesvirus 4 | [46] |
| Human (Healthy) | Longitudinal metagenomics analysis (Ireland) of fecal viruses N = 6 (3 Males, 3 Females) | Taxa identified: ↑ Virulent crAss-like and Microviridae bacteriophages | [18] |
| Uncultured viral community from human feces. N = 1 | Taxa identified: Bacteriophages A118 of Listeria monocytogenes, E125 of Burkholderia thailandensis, and bIL285 of Lactococcus lactis | [47] | |
| Analysis of the RNA viruses (N = 2) | Pepper mild mottle virus = 109 virions/gram of dry fecal matter, ↑ RNA viruses | [48] | |
| Virome of the ELDERMET | First-ever in elders (>65 yrs) | Taxa identified:Gokushovirinae (Microviridae) | [49] |
| Diet variations | sequencing (N = 6) high-fat/low-fiber diet | High fat to low fiber: Siphoviridae (18%), 686 (10%) to Myoviridae, 344 (4.8%) to Podoviridae, 68 (0.9%) to Microviridae, and 0.4% others | [50] |
| Human Virome (IBD) | Virus-like particle preparations on the rectal mucosa (N = 167, (UC = 91; HC = 76)). (Chinese study) | UC to HC = ↓ mucosal Caudovirales diversity, richness and evenness relative to HC | [51] |
| metagenomics sequencing of stool filtrates using the Roche 454 platform (UK) | The viromes of CD and UC patients were disease- and cohort-specific. ↑ Caudovirales bacteriophages compared to HC | [52] | |
| T2D vs. HC | Type II diabetes (T2D) patients (n = 71) and normal Chinese adults (HC) n = 74)). | T2D to HC: ↑ phages (Siphoviridae (55.3 ± 9.8%) Myoviridae (21.7 ± 9.9%), Podoviridae (10.6 ± 8.4%)) | [53] |
| HIV study | (N = 122, untreated HIV = 42, HIV ART= 40, HIV uninfected= 40) | HIV to uninfected: ↑ Anelloviridae, Adenoviruses | [54] |
| Animal | |||
| Gorilla simian immunodeficiency virus infection (SIV). | (N = 22, SIV = 11, HC = 11) | SIV to HC: ↑ Herpesviridae and Reoviridae Identified: Siphoviridae, Myoviridae and Podoviridae | [55] |
| Rodents | N = 314 wild rodent | Taxa identified: Parvovirus, Dicistrovirus, Iflavirus, and Iridovirus | [56] |
| viral RNA and DNA in the feces of 105 wild rodents | Taxa identified:Circoviridae, Picobirnaviridae, Picornaviridae, Astroviridae, Parvoviridae, Papillomaviridae, Adenoviridae, and Coronaviridae. | [57] | |
| viral content in rat fecal matter (N = 29) | Picornaviridae | [58] | |
| Mouse | N = 416 mice | Taxa identified: Parvovirinae, Chapparvovirus, Polyomavirus, Astroviruses, Sapovirus, Picornavirus | [59] |
| Mouse model of IBD | C57BL6/J mice (HC) and C57BL6/J Rag1-/- mice (IBD induced) | IBD to HC= ↑ Spounaviridae, ↓ Clostridiales phages | [60] |
| Gulf War illness (GWI) mouse model (IBD) | (N = 22, HC = 11, GWI = 11) | GWI mice to HC = ↓ Microviridae bacteriophages, ↑ Siphoviridae and Myoviridae bacteriophages | [61] |
| Gnotobiotic mouse model of phage-bacterial host dynamics | (N = 5 per group). | T7 phages are undetectable for 1 wk in germ-free animals before they rise in abundance after gavage of a bacterial host. | [62] |
| Phages | ds DNA | ss DNA | ds RNA | ss RNA |
|---|---|---|---|---|
| Microviridae | Papillomaviridae | Circoviridae | Reoviridae | Retroviridae |
| Podoviridae | Polyomaviridae | Anelloviridae | Togaviridae | |
| Siphoviridae | Poxviridae | Astroviridae | ||
| Adenoviridae | Virgaviridae | |||
| Iridoviridae | Caliciviridae | |||
| Marseilleviridae |
| Model | Study Details | Findings | Ref. |
|---|---|---|---|
| Infant IBD | N = 4, Infant UC = 3, HC = 1 (22 to 30 FMT treatments | UC to HC: ↑ Siphoviridae | [72] |
| Adult IBD | N = 15, UC= 9, HC = 8 | UC to HC: ↓ DNA phage, richness of donor viromes ≠ outcome of therapy Most abundant: Anelloviridae, Circoviridae, Picobirnaviridae and Virgaviridae | [81] |
| Clostridium difficile infection (CDI) | N = 44, CDI= 24, HC = 20 | CDI to HC: ↑ Caudovirales (may play a role in FMT efficacy in CDI) | [73] |
| Recurrent CDI (rCDI) 1-year follow-up | rCDI = 14; donors (D) = 3 | rCDI to D: ↑ Caudovirales, Anelloviridae ↓ Microviridae | [74] |
| Model type | Study details | Findings | Ref |
|---|---|---|---|
| Human | |||
| Mycobiome of Human microbiome project (HMP) | N = 317 | Taxa identified: Saccharomyces, Malassezia, and Candida. (↓ Mycobiome diversity) | [20,101] |
| Mother/offspring (prospective cohort) | N = 298 pairs (mothers and offspring) | From Mothers to off springs: ↑ Debaryomyces hansenii (breast-feeding) ↑ S. cerevisiae (after weaning) | [101] |
| The New Zealand human healthy gut mycobiome | N = 21 healthy, non-obese (age: 18–65 yr) | Taxa identified: Candida albicans, Candida parapsilosis, and S. cerevisiae. New species identified: C. bracarensis, Coniochaeta hoffmannii, Hanseniaspora pseudoguilliermondii, Aspergillus foetidus, A. tubingensis, and Paecilomyces dactylethromorphus | [102] |
| Healthy Aging study: Gut mycobiome of elderly Danish people (Age: 65–81 yr) | N = 99 (age:65 to 81 yr) | Phyla: Ascomycota, Basidiomycota and Zygomycota Genera: Penicillium, Candida, and Aspergillus | [103] |
| Mycobiota among Eutrophic, overweight, and obese | N = 72, Eutrophic = 24, Overweight = 24, Obese = 24). | Eutrophic: Zygomycota and Basidiomycota, Overweight: Zygomycota and Basidiomycota, Obese: Zygomycota, Basidiomycota and Syncephalastrum sp. (Zygomycota) Taxa identified include: ↑ Ascomycota (species): Paecilomyces sp., Penicillium sp., Candida sp., Aspergillus sp., Fonsecaea sp., and Geotrichum sp. ↑ Basidiomycota (species): Trichosporon sp. and Rhodotorula sp. ↑ Zygomycota (species) Rhizopus sp. and Mucor sp. | [104] |
| Healthy Japanese gut Mycobiota | N = 14 | Taxa identified: ↑ Candida and Saccharomyces | [21] |
| Intestinal mycobiome of patients with irritable bowel syndrome (IBS). | N = 57, HC = 18 healthy IBS = 39 | S. cerevisiae and C albicans identified in all samples/groups | [105] |
| Animal | |||
| Rat model of visceral hypersensitivity | N = 6 | S. cerevisiae and C. albicans in all samples. | [105] |
| Bat | N = 14 | ↑ Ascomycota and Basidiomycota | [106] |
| Dog | N = 19, HC= 12, acute diarrhea (AD)= 7 | Ascomycota (HC: 97.9% and AD: 98.2%) and Basidiomycota (HC: 1.0%, AD: 0.5%) | [107] |
| Mouse | Pancreatic ductal adenocarcinoma (PDA) and C57BL/6 mice (HC). | PDA to HC: ↑ Malassezia species promote PDA | [108] |
| Tibetan macaque | n.a | Taxa identified:Zygomycota, Chytridiomycota, Glomeromycota and Rozellomycota | [109] |
| Model type | Study details | Findings | Ref |
|---|---|---|---|
| Human | |||
| Healthy infants | N = 15, Cesarean section-delivered (CSD) = 8, Vaginally-derived(VD) = 7 | CSD to VD: ↑ Methanosphaera spp. | [121] |
| Infants/mothers (healthy) | N = 8 (4 twin pairs) | Lipothrixviridae | [43] |
| Obese children | N = 20 (HC = 10, Obese = 10) | Obese to HC: ↑ Methanobrevibacter spp. | [46] |
| N = 476 | Obese to HC: ↑ M. smithii | [122] | |
| Healthy adults | N = 8 | M. smithii (99–100%) | [123] |
| N = 15 (Finnish) | Taxa identified: Methanobrevibacter–specific | [124] | |
| Population-based | Belgian Flemish Gut Flora Project (FGFP; discovery cohort; N = 1106) and the Dutch LifeLines-DEEP study (LLDeep; replication; N = 1135) | ↑ Methanobrevibacter spp. in all samples | [125] |
| Aging study | N = 500 | Taxa identified: M. smithii Methanosphaera stadtmanae Methanomassiliicoccus luminyensis | [126] |
| Archaea of ELDERMET study | N = 371 | Taxa identified: Methanomassiliicoccales | [127] |
| Type 2 diabetes | N = 49 HC= 19, New (type 2) = 14, Known (type 2)= 16 | NGTs to Known = ↓ Methanobrevibacter | [22] |
| Human IBD | N = 58, HC = 29, IBD = 29 | IBD to HC: ↑ M. stadtmanae | [128] |
| N = 108, HC= 47, IBD = 61 | IBD to HC: ↓ M. smithii | [129] | |
| Animal | |||
| Human/ape study | Humans (N = 10) Apes: Pan troglodytes (chimpanzee = 14), Pan paniscus (bonobo = 18), Gorilla gorilla (gorilla = 20), and Pongo pygmaeus (orangutan = 8) | Taxa identified: M. smithii and M. stadtmanae in all samples (more in humans) | [24] |
| Rabbit cecal archaea | N = 40 | Taxa identified: Methanobrevibacter and Methanosphaera spp. in all samples | [130] |
| Model Type | Study Details | Findings | Ref. |
|---|---|---|---|
| Rat model (2, 4, 6 Trinitrobenzene sulphonic acid (TNBS) induced colitis model | N = 24: HC = 6, Schistosoma mansoni group = 6, TNBS group = 6 and S. mansoni + TNBS group= 6 | S. mansoni group: ↑ IL-2, IL-4 TNBS group: ↑ IL-2 ↑ T helper 1 (Th1) S. mansoni plus TNBS group: ↑ Th2 ↓ Th 1 (↓ Inflammation) Concurrent infection with S. mansoni significantly attenuates TNBS induced colitis in the rats. | [138] |
| Rhesus macaques with idiopathic chronic diarrhea (ICD) w/o T suis infection | N = 7, ICD = 5 ICD with T suis = 5, HC = 2 | ICD group = ↓ Mucosal bacterial diversity, ↓ Th2 ICD with T suis = ↑ Mucosal bacterial diversity (Cyanobacteria), ↑ Th2 | [139] |
| Human (Tetanus toxoid (TT) and S mansoni infection) | TT + S. mansoni = 11 HC= 5 | TT + S. mansoni group: ↓ Interferon gamma (IFN-ɤ), ↑ Th 2 HC: ↑ IFN-ɤ ↓ Th1 | [140] |
| Human (helminthic ova in the treatment of active IBD) | N = 4 (IBD), 2500 live Trichuris suis eggs/12 weeks/subject | IBD: ↑ Inflammation: ↑ Th1 T. suis treated: ↓ Inflammation: ↑ Th2 and ↓ Th1 | [141] |
| Human (RCT T suis therapy for ulcerative colitis (UC)) | N = 54, Therapy group= 30, HC = 24 | Therapy group =↓ UC disease activity index (DAI) | [137] |
| Human (RCT T suis therapy for Crohn’s disease (CD)) | N = 36, Treated group = 27, HC = 9 | T. suis therapy was well tolerated | [142] |
| Human (Necator americanus therapy for CD) | N = 18, (inoculation) | CD: ↑ DAI N. americanus treated: ↓ CD (DAI) | [143] |
| Human (T suis therapy multiple sclerosis) | N = 5 | Multiple sclerosis: ↑ Inflammation T. suis treated: ↑ IL-4, ↑ IL-10 (↓ Inflammation = ↑ Th 2?) | [144] |
| Microbial Constituent | Response Type | Immune Cells/Expressions | Cytokines | Initiation |
|---|---|---|---|---|
| Bacteria (intracellular): Systemic commensals, Proteobacteria, pathogens | Inflammatory response (IR) 1 | T helper 1 (Th 1) cells | Interferon gamma (IFN-ɤ), Tumor necrosis factor (TNF-α) | Microbe-associated molecular patterns (Pro-inflammatory) |
| Virus | IR 1 | CD4, CD8 T cells | IFN-α/β, IFN-λ | |
| Segmented filamentous bacteria (extracellular), Fungi | IR 2 | Th 17 cells | Interleukins (IL) 17A, IL-22 | Mucosal epithelial cells |
| Helminths | IR 2 | Th 2 cells | IFN-ɤ | GATA 3 |
| Archaea ((Methanomassiliicoccus luminyensis, Methanosphaera stadtmanae and M. smithii)) | Inflammatory/regulatory response | T cells- CD86, CD197 | Monocyte-derived dendritic cells (MODC), type 1 IFN | Mucosal epithelial cells |
| Clostridia, Bacteroides fragilis, archaea and helminths | Regulatory response | T regulatory cells (Foxp3 + Tregs) | IL-10, Transforming growth factor beta (TGF-β) | Resolution of IR 1, 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vemuri, R.; Shankar, E.M.; Chieppa, M.; Eri, R.; Kavanagh, K. Beyond Just Bacteria: Functional Biomes in the Gut Ecosystem Including Virome, Mycobiome, Archaeome and Helminths. Microorganisms 2020, 8, 483. https://doi.org/10.3390/microorganisms8040483
Vemuri R, Shankar EM, Chieppa M, Eri R, Kavanagh K. Beyond Just Bacteria: Functional Biomes in the Gut Ecosystem Including Virome, Mycobiome, Archaeome and Helminths. Microorganisms. 2020; 8(4):483. https://doi.org/10.3390/microorganisms8040483
Chicago/Turabian StyleVemuri, Ravichandra, Esaki M. Shankar, Marcello Chieppa, Rajaraman Eri, and Kylie Kavanagh. 2020. "Beyond Just Bacteria: Functional Biomes in the Gut Ecosystem Including Virome, Mycobiome, Archaeome and Helminths" Microorganisms 8, no. 4: 483. https://doi.org/10.3390/microorganisms8040483
APA StyleVemuri, R., Shankar, E. M., Chieppa, M., Eri, R., & Kavanagh, K. (2020). Beyond Just Bacteria: Functional Biomes in the Gut Ecosystem Including Virome, Mycobiome, Archaeome and Helminths. Microorganisms, 8(4), 483. https://doi.org/10.3390/microorganisms8040483