The Role of Petrimonas mucosa ING2-E5AT in Mesophilic Biogas Reactor Systems as Deduced from Multiomics Analyses

 , , , ,
, , , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Strain Cultivation and DNA Isolation
2.2. PacBio Library Preparation, Sequencing, and Genome Assembly
2.3. Reconstruction of Metabolic Pathways
2.4. Fragment Recruitment
2.5. Petrimonas Transcriptional Profile as Deduced from Public Transcriptome Datasets
2.6. Gene Expression Analysis of Petrimonas Species Based on Database Metaproteome Datasets
2.7. Nucleotide Sequence Accession Number
3. Results and Discussion
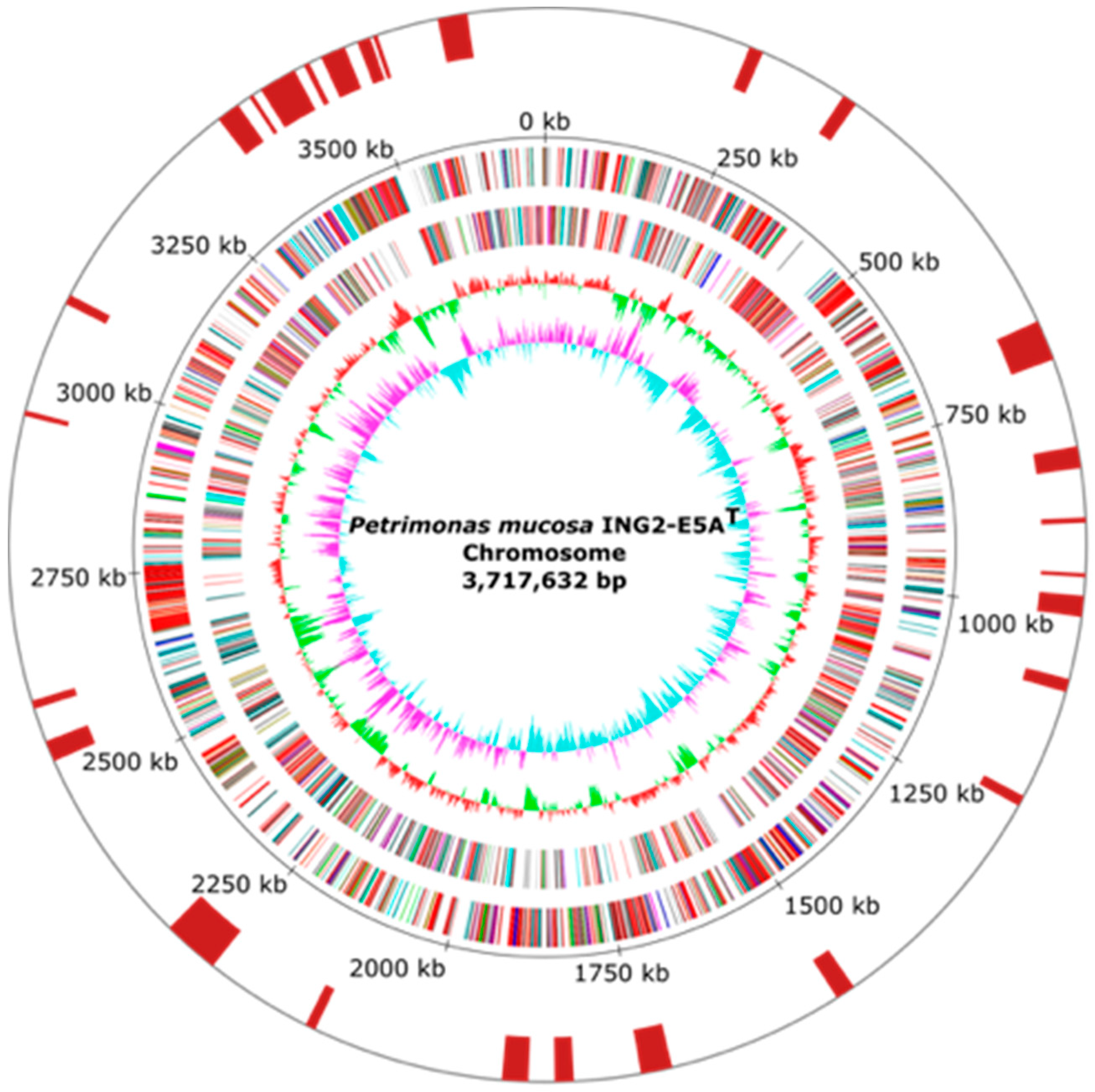
3.1. General Features of Petrimonas mucosa ING2-E5AT Genome
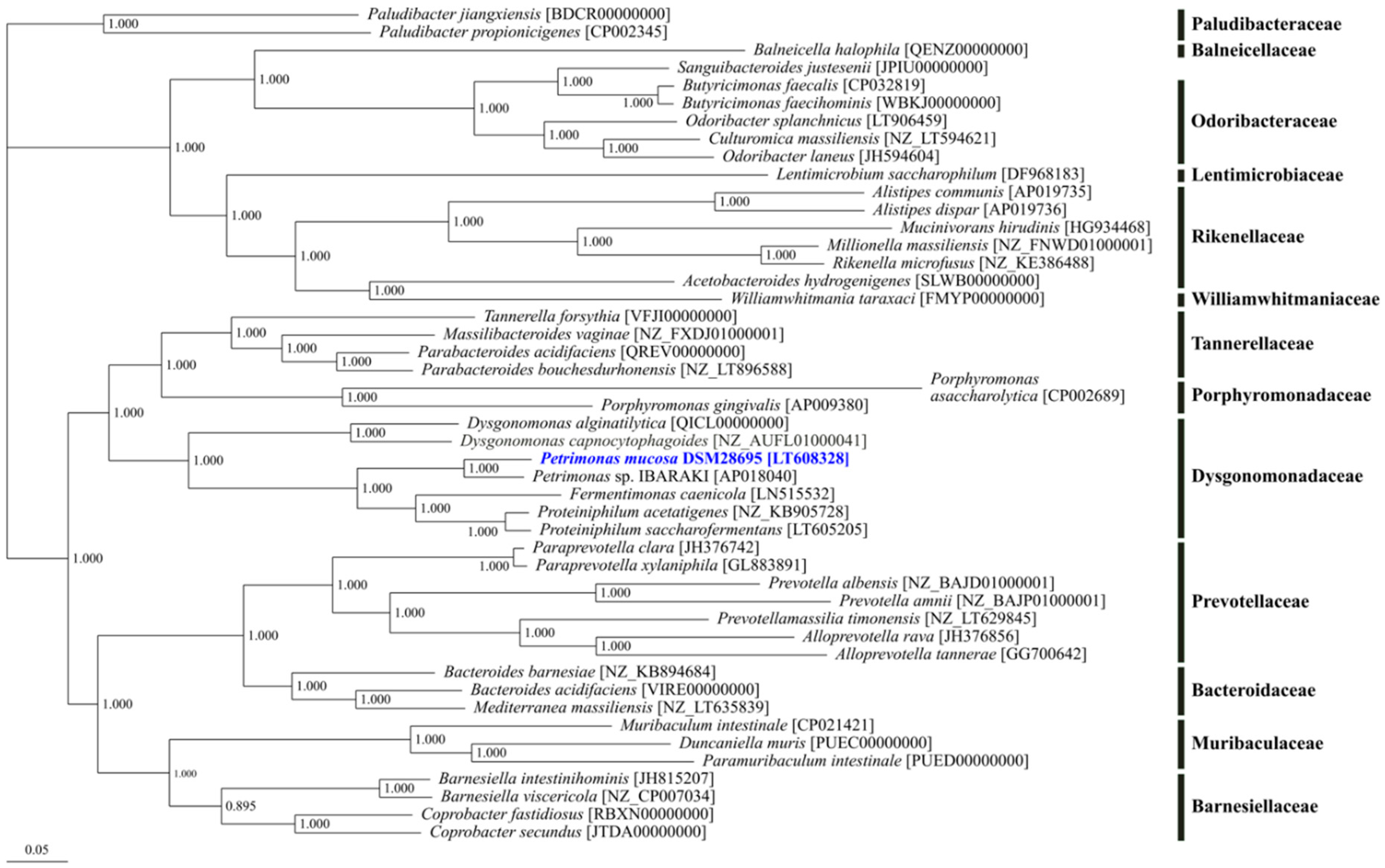
3.2. Phylogenetic Classification of Petrimonas mucosa ING2-E5AT
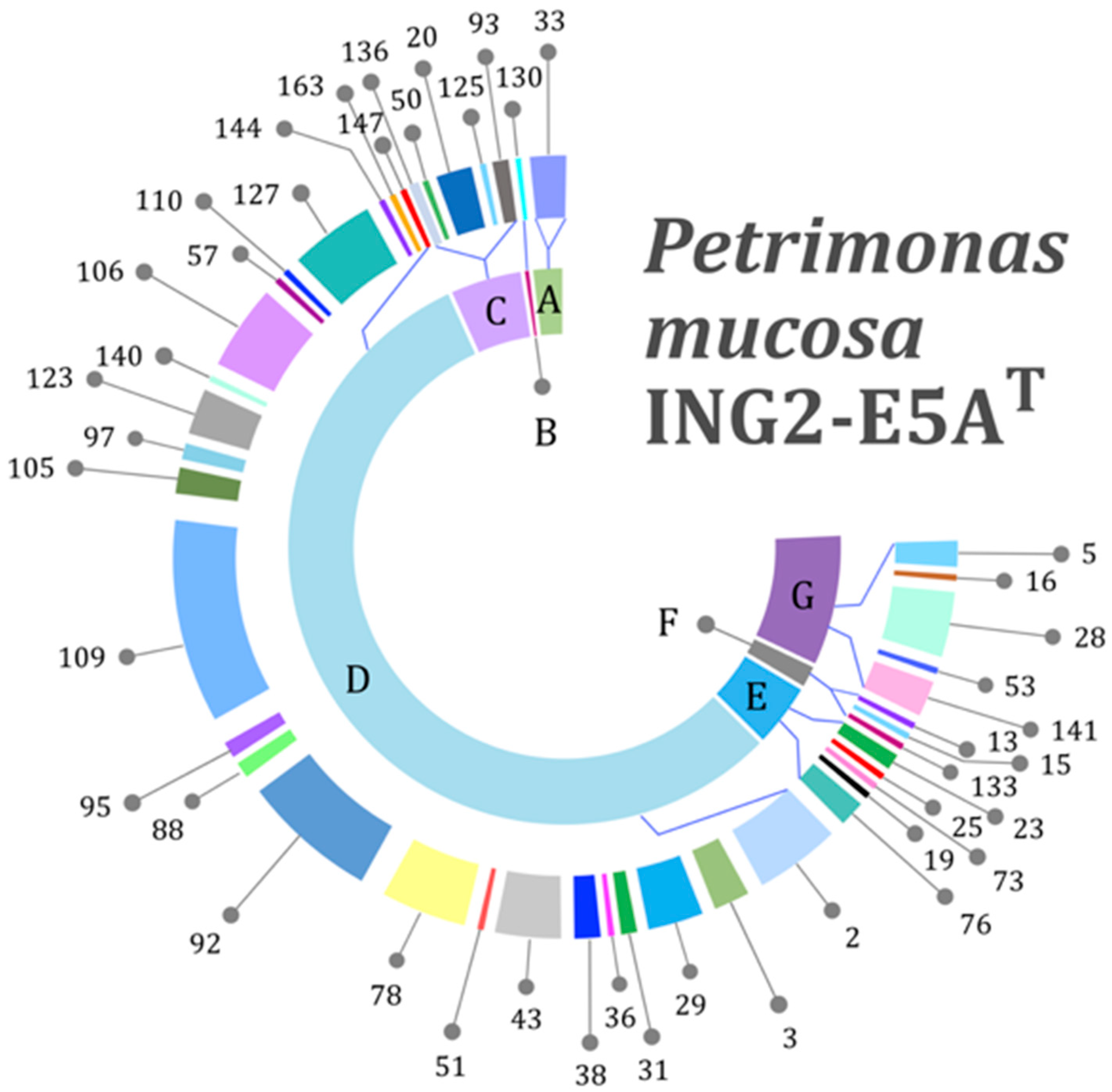
3.3. Genes Encoding Carbohydrate-Active Enzymes
3.4. Genes Encoding Polysaccharide Utilization Loci (PUL)
3.5. Genes Encoding Enzymes of the Central Fermentation Metabolism
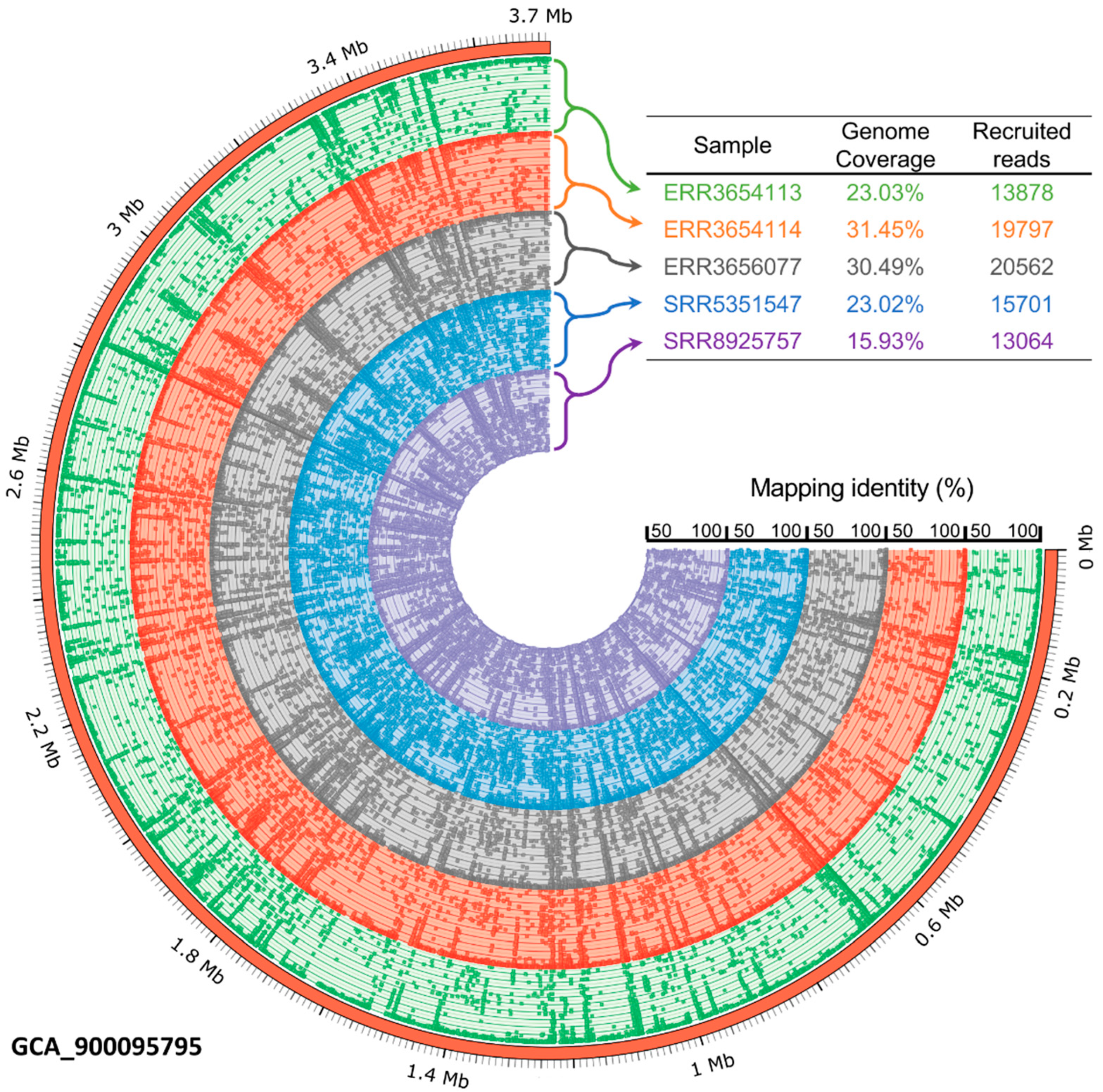
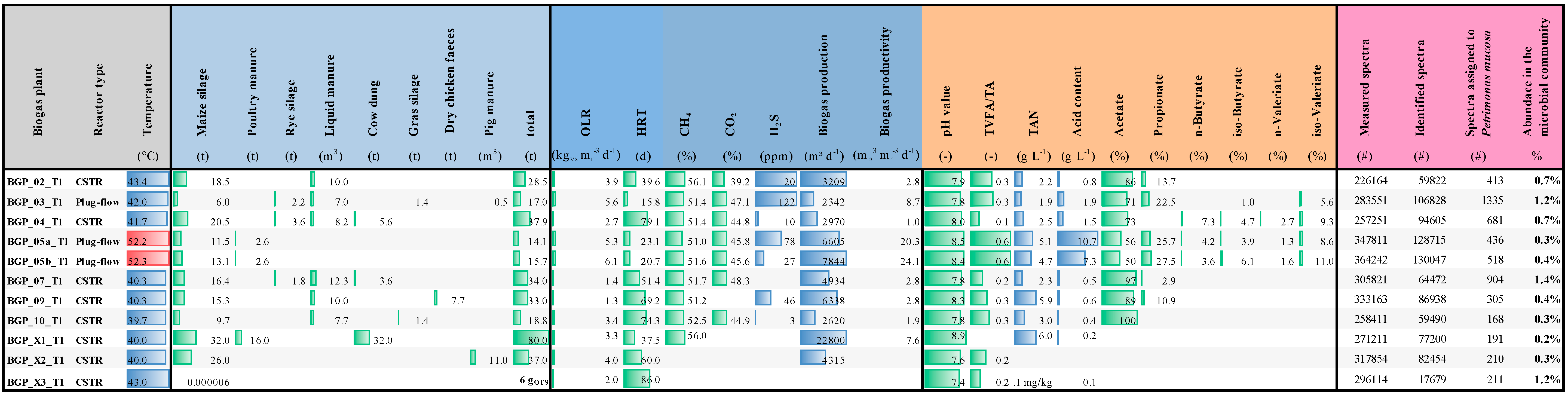
3.6. The Occurrence of Petrimonas Relatives in Biogas-Producing Microbial Communities as Deduced from Publicly Available Metagenome Data
3.7. The Transcriptional Profile of Petrimonas Species
3.8. Metaproteome Analysis Revealed the Presence of Petrimonas spp. Proteins in Various Biogas-Producing Communities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Venkiteshwaran, K.; Bocher, B.; Maki, J.; Zitomer, D. Relating Anaerobic Digestion Microbial Community and Process Function. Microbiol. Insights 2015, 8, 37–44. [Google Scholar] [PubMed]
- Schnürer, A. Biogas Production: Microbiology and Technology. Adv. Biochem. Eng. Biotechnol. 2016, 156, 195–234. [Google Scholar] [PubMed]
- Westerholm, M.; Dolfing, J.; Schnürer, A. Growth Characteristics and Thermodynamics of Syntrophic Acetate Oxidizers. Environ. Sci. Technol. 2019, 53, 5512–5520. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, R.; Saady, N.M.C.; Torrijos, M.; Thanikal, J.V.; Hung, Y.-T. Sustainable Agro-Food Industrial Wastewater Treatment Using High Rate Anaerobic Process. Water 2013, 5, 292. [Google Scholar] [CrossRef]
- Niu, Q.; Hojo, T.; Qiao, W.; Qiang, H.; Li, Y.-Y. Characterization of Methanogenesis, Acidogenesis and Hydrolysis in Thermophilic Methane Fermentation of Chicken Manure. Chem. Eng. J. 2014, 244, 587–596. [Google Scholar] [CrossRef]
- Lv, Z.; Wu, X.; Zhou, B.; Wang, Y.; Sun, Y.; Wang, Y.; Chen, Z.; Zhang, J. Effect of One Step Temperature Increment from Mesophilic to Thermophilic Anaerobic Digestion on the Linked Pattern between Bacterial and Methanogenic Communities. Bioresour. Technol. 2019, 292, 121968. [Google Scholar] [CrossRef]
- He, Q.; Li, L.; Zhao, X.; Qu, L.; Wu, D.; Peng, X. Investigation of Foaming Causes in Three Mesophilic Food Waste Digesters: Reactor Performance and Microbial Analysis. Sci. Rep. 2017, 7, 13701. [Google Scholar] [CrossRef]
- Yenigün, O.; Demirel, B. Ammonia Inhibition in Anaerobic Digestion: A Review. Process. Biochem. 2013, 48, 901–911. [Google Scholar] [CrossRef]
- Maus, I.; Klocke, M.; Derenkó, J.; Stolze, Y.; Beckstette, M.; Jost, C.; Wibberg, D.; Blom, J.; Henke, C.; Willenbücher, K.; et al. Impact of Process Temperature and Organic Loading Rate on Cellulolytic/Hydrolytic Biofilm Microbiomes during Biomethanation of Ryegrass Silage Revealed by Genome-Centered Metagenomics and Metatranscriptomics. Environ. Microbiome 2020, 15, 7. [Google Scholar] [CrossRef]
- He, Q.; Li, L.; Peng, X. Early Warning Indicators and Microbial Mechanisms for Process Failure Due to Organic Overloading in Food Waste Digesters. J. Environ. Eng. 2017, 143, 04017077. [Google Scholar] [CrossRef]
- Rajagopal, R.; Massé, D.I.; Singh, G. A Critical Review on Inhibition of Anaerobic Digestion Process by Excess Ammonia. Bioresour. Technol. 2013, 143, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Campanaro, S.; Treu, L.; Kougias, P.G.; De Francisci, D.; Valle, G.; Angelidaki, I. Metagenomic Analysis and Functional Characterization of the Biogas Microbiome Using High Throughput Shotgun Sequencing and a Novel Binning Strategy. Biotechnol. Biofuels 2016, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Sun, L.; Westerholm, M.; Schnürer, A. Bacterial Community Composition and Fhs Profiles of Low- and High-Ammonia Biogas Digesters Reveal Novel Syntrophic Acetate-Oxidising Bacteria. Biotechnol. Biofuels 2016, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Treu, L.; Kougias, P.G.; Campanaro, S.; Bassani, I.; Angelidaki, I. Deeper Insight into the Structure of the Anaerobic Digestion Microbial Community; the Biogas Microbiome Database Is Expanded with 157 New Genomes. Bioresour. Technol. 2016, 216, 260–266. [Google Scholar] [CrossRef]
- Stolze, Y.; Bremges, A.; Maus, I.; Pühler, A.; Sczyrba, A.; Schlüter, A. Targeted in Situ Metatranscriptomics for Selected Taxa from Mesophilic and Thermophilic Biogas Plants. Microb. Biotechnol. 2018, 11, 667–679. [Google Scholar] [CrossRef]
- Maus, I.; Koeck, D.E.; Cibis, K.G.; Hahnke, S.; Kim, Y.S.; Langer, T.; Kreubel, J.; Erhard, M.; Bremges, A.; Off, S.; et al. Unraveling the Microbiome of a Thermophilic Biogas Plant by Metagenome and Metatranscriptome Analysis Complemented by Characterization of Bacterial and Archaeal Isolates. Biotechnol. Biofuels 2016, 9, 171. [Google Scholar] [CrossRef]
- Fischer, M.A.; Güllert, S.; Refai, S.; Künzel, S.; Deppenmeier, U.; Streit, W.R.; Schmitz, R.A. Long-term Investigation of Microbial Community Composition and Transcription Patterns in a Biogas Plant Undergoing Ammonia Crisis. Microb. Biotechnol. 2018, 12, 305–323. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G.; et al. The Anaerobic Digestion Microbiome: New Insights from the Biogas Microbiome by Comprehensive Genome-Resolved Metagenomics of Nearly 1600 Species Originating from Multiple Anaerobic Digesters. Biotechnol. Biofuels 2020, 13, 25. [Google Scholar] [CrossRef]
- Zhang, J.; Loh, K.-C.; Lee, J.; Wang, C.-H.; Dai, Y.; Wah Tong, Y. Three-Stage Anaerobic Co-Digestion of Food Waste and Horse Manure. Sci. Rep. 2017, 7, 1269. [Google Scholar] [CrossRef]
- Sun, L.; Pope, P.B.; Eijsink, V.G.H.; Schnürer, A. Characterization of Microbial Community Structure during Continuous Anaerobic Digestion of Straw and Cow Manure. Microb. Biotechnol. 2015, 8, 815–827. [Google Scholar] [CrossRef]
- Moset, V.; Poulsen, M.; Wahid, R.; Højberg, O.; Møller, H.B. Mesophilic versus Thermophilic Anaerobic Digestion of Cattle Manure: Methane Productivity and Microbial Ecology. Microb. Biotechnol. 2015, 8, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Goux, X.; Calusinska, M.; Lemaigre, S.; Marynowska, M.; Klocke, M.; Udelhoven, T.; Benizri, E.; Delfosse, P. Microbial Community Dynamics in Replicate Anaerobic Digesters Exposed Sequentially to Increasing Organic Loading Rate, Acidosis, and Process Recovery. Biotechnol. Biofuels 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Hofstad, T.; Olsen, I.; Eribe, E.R.; Falsen, E.; Collins, M.D.; Lawson, P.A. Dysgonomonas Gen. Nov. to Accommodate Dysgonomonas Gadei Sp. Nov., an Organism Isolated from a Human Gall Bladder, and Dysgonomonas Capnocytophagoides (Formerly CDC Group DF-3). Int. J. Syst. Evol. Microbiol. 2000, 50, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Dong, X. Proteiniphilum Acetatigenes Gen. Nov., Sp. Nov., from a UASB Reactor Treating Brewery Wastewater. Int. J. Syst. Evol. Microbiol. 2005, 55, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Hahnke, S.; Langer, T.; Koeck, D.E.; Klocke, M. Description of Proteiniphilum Saccharofermentans Sp. Nov., Petrimonas Mucosa Sp. Nov. and Fermentimonas Caenicola Gen. Nov., Sp. Nov., Isolated from Mesophilic Laboratory-Scale Biogas Reactors, and Emended Description of the Genus Proteiniphilum. Int. J. Syst. Evol. Microbiol. 2016, 66, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, A.; Tindall, B.J.; Bardin, V.; Blanchet, D.; Jeanthon, C. Petrimonas Sulfuriphila Gen. Nov., Sp. Nov., a Mesophilic Fermentative Bacterium Isolated from a Biodegraded Oil Reservoir. Int. J. Syst. Evol. Microbiol. 2005, 55, 1113–1121. [Google Scholar] [CrossRef]
- García-López, M.; Meier-Kolthoff, J.P.; Tindall, B.J.; Gronow, S.; Woyke, T.; Kyrpides, N.C.; Hahnke, R.L.; Göker, M. Analysis of 1000 Type-Strain Genomes Improves Taxonomic Classification of Bacteroidetes. Front. Microbiol. 2019, 10, 2083. [Google Scholar] [CrossRef]
- Sakamoto, M. The Family Porphyromonadaceae. In The Prokaryotes: Other Major Lineages of Bacteria and the Archaea; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 811–824. [Google Scholar]
- Tomazetto, G.; Hahnke, S.; Wibberg, D.; Pühler, A.; Klocke, M.; Schlüter, A. Proteiniphilum Saccharofermentans Str. M3/6T Isolated from a Laboratory Biogas Reactor Is Versatile in Polysaccharide and Oligopeptide Utilization as Deduced from Genome-Based Metabolic Reconstructions. Biotechnol. Rep. 2018, 18, e00254. [Google Scholar] [CrossRef]
- Koeck, D.E.; Zverlov, V.V.; Liebl, W.; Schwarz, W.H. Comparative Genotyping of Clostridium Thermocellum Strains Isolated from Biogas Plants: Genetic Markers and Characterization of Cellulolytic Potential. Syst. Appl. Microbiol. 2014, 37, 311–319. [Google Scholar] [CrossRef]
- Cibis, K.G.; Gneipel, A.; König, H. Isolation of Acetic, Propionic and Butyric Acid-Forming Bacteria from Biogas Plants. J. Biotechnol. 2016, 220, 51–63. [Google Scholar] [CrossRef]
- Tejerizo, G.T.; Kim, Y.S.; Maus, I.; Wibberg, D.; Winkler, A.; Off, S.; Pühler, A.; Scherer, P.; Schlüter, A. Genome Sequence of Methanobacterium Congolense Strain Buetzberg, a Hydrogenotrophic, Methanogenic Archaeon, Isolated from a Mesophilic Industrial-Scale Biogas Plant Utilizing Bio-Waste. J. Biotechnol. 2017, 247, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Pechtl, A.; Rückert, C.; Maus, I.; Koeck, D.E.; Trushina, N.; Kornberger, P.; Schwarz, W.H.; Schlüter, A.; Liebl, W.; Zverlov, V.V. Complete Genome Sequence of the Novel Cellulolytic, Anaerobic, Thermophilic Bacterium Herbivorax Saccincola Type Strain GGR1, Isolated from a Lab Scale Biogas Reactor as Established by Illumina and Nanopore MinION Sequencing. Genome Announc. 2018, 6, e01493-17. [Google Scholar] [CrossRef] [PubMed]
- Maus, I.; Bremges, A.; Stolze, Y.; Hahnke, S.; Cibis, K.G.; Koeck, D.E.; Kim, Y.S.; Kreubel, J.; Hassa, J.; Wibberg, D.; et al. Genomics and Prevalence of Bacterial and Archaeal Isolates from Biogas-Producing Microbiomes. Biotechnol. Biofuels 2017, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Nelkner, J.; Henke, C.; Lin, T.W.; Pätzold, W.; Hassa, J.; Jaenicke, S.; Grosch, R.; Pühler, A.; Sczyrba, A.; Schlüter, A. Effect of Long-Term Farming Practices on Agricultural Soil Microbiome Members Represented by Metagenomically Assembled Genomes (MAGs) and Their Predicted Plant-Beneficial Genes. Genes 2019, 10, 424. [Google Scholar] [CrossRef]
- Meyer, F.; Goesmann, A.; McHardy, A.C.; Bartels, D.; Bekel, T.; Clausen, J.; Kalinowski, J.; Linke, B.; Rupp, O.; Giegerich, R.; et al. GenDB—An Open Source Genome Annotation System for Prokaryote Genomes. Nucleic Acids Res. 2003, 31, 2187–2195. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Dhillon, B.K.; Laird, M.R.; Shay, J.A.; Winsor, G.L.; Lo, R.; Nizam, F.; Pereira, S.K.; Waglechner, N.; McArthur, A.G.; Langille, M.G.I.; et al. IslandViewer 3: More Flexible, Interactive Genomic Island Discovery, Visualization and Analysis. Nucleic Acids Res. 2015, 43, W104–W108. [Google Scholar] [CrossRef]
- Blom, J.; Kreis, J.; Spänig, S.; Juhre, T.; Bertelli, C.; Ernst, C.; Goesmann, A. EDGAR 2.0: An Enhanced Software Platform for Comparative Gene Content Analyses. Nucleic Acids Res. 2016, 44, W22–W28. [Google Scholar] [CrossRef]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. DbCAN: A Web Resource for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, D.; Pedersen, C.N.S.; Greening, C. HydDB: A Web Tool for Hydrogenase Classification and Analysis. Sci. Rep. 2016, 6, 34212. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Krüger, J.; Sczyrba, A. Analyzing Large Scale Genomic Data on the Cloud with Sparkhit. Bioinformatics 2018, 34, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Zhu, Z.; Fu, L.; Wu, S.; Li, W. FR-HIT, a Very Fast Program to Recruit Metagenomic Reads to Homologous Reference Genomes. Bioinformatics 2011, 27, 1704–1705. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Trim Galore. Available online: https://www.psc.edu/user-resources/software/trim-galore (accessed on 20 October 2020).
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. RnaSPAdes: A de Novo Transcriptome Assembler and Its Application to RNA-Seq Data. Gigascience 2019, 8, giz100. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Siewert, C.; Kohrs, F.; Greve, J.; Maus, I.; Klang, J.; Klocke, M.; Heiermann, M.; Hoffmann, M.; et al. Metaproteome Analysis Reveals That Syntrophy, Competition, and Phage-Host Interaction Shape Microbial Communities in Biogas Plants. Microbiome 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Beavis, R.C. TANDEM: Matching Proteins with Tandem Mass Spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Markey, S.P.; Kowalak, J.A.; Wagner, L.; Xu, M.; Maynard, D.M.; Yang, X.; Shi, W.; Bryant, S.H. Open Mass Spectrometry Search Algorithm. J. Proteome Res. 2004, 3, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Heyer, R.; Schallert, K.; Büdel, A.; Zoun, R.; Dorl, S.; Behne, A.; Kohrs, F.; Püttker, S.; Siewert, C.; Muth, T.; et al. A Robust and Universal Metaproteomics Workflow for Research Studies and Routine Diagnostics Within 24 h Using Phenol Extraction, FASP Digest, and the MetaProteomeAnalyzer. Front. Microbiol. 2019, 10, 1883. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, A.; Zakrzewski, M.; Schlüter, A.; Schönberg, M.; Szczepanowski, R.; Goesmann, A.; Pühler, A.; Klocke, M. Characterization of Microbial Biofilms in a Thermophilic Biogas System by High-Throughput Metagenome Sequencing. FEMS Microbiol. Ecol. 2012, 79, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Hanreich, A.; Schimpf, U.; Zakrzewski, M.; Schlüter, A.; Benndorf, D.; Heyer, R.; Rapp, E.; Pühler, A.; Reichl, U.; Klocke, M. Metagenome and Metaproteome Analyses of Microbial Communities in Mesophilic Biogas-Producing Anaerobic Batch Fermentations Indicate Concerted Plant Carbohydrate Degradation. Syst. Appl. Microbiol. 2013, 36, 330–338. [Google Scholar] [CrossRef]
- Stolze, Y.; Bremges, A.; Rumming, M.; Henke, C.; Maus, I.; Pühler, A.; Sczyrba, A.; Schlüter, A. Identification and Genome Reconstruction of Abundant Distinct Taxa in Microbiomes from One Thermophilic and Three Mesophilic Production-Scale Biogas Plants. Biotechnol. Biofuels 2016, 9, 156. [Google Scholar] [CrossRef]
- Schlüter, A.; Bekel, T.; Diaz, N.N.; Dondrup, M.; Eichenlaub, R.; Gartemann, K.-H.; Krahn, I.; Krause, L.; Krömeke, H.; Kruse, O.; et al. The Metagenome of a Biogas-Producing Microbial Community of a Production-Scale Biogas Plant Fermenter Analysed by the 454-Pyrosequencing Technology. J. Biotechnol. 2008, 136, 77–90. [Google Scholar] [CrossRef]
- Pikuta, E.V.; Lyu, Z.; Hoover, R.B.; Liu, Y.; Patel, N.B.; Busse, H.J.; Lawson, P.A. Williamwhitmania Taraxaci Gen. Nov., Sp. Nov., a Proteolytic Anaerobe with a Novel Type of Cytology from Lake Untersee in Antarctica, Description of Williamwhitmaniaceae Fam. Nov., and Emendation of the Order Bacteroidales Krieg 2012. Int. J. Syst. Evol. Microbiol. 2017, 67, 4132–4145. [Google Scholar] [CrossRef]
- Ormerod, K.L.; Wood, D.L.A.; Lachner, N.; Gellatly, S.L.; Daly, J.N.; Parsons, J.D.; Dal’Molin, C.G.O.; Palfreyman, R.W.; Nielsen, L.K.; Cooper, M.A.; et al. Genomic Characterization of the Uncultured Bacteroidales Family S24-7 Inhabiting the Guts of Homeothermic Animals. Microbiome 2016, 4, 36. [Google Scholar] [CrossRef]
- CAZypedia Consortium. Ten Years of CAZypedia: A Living Encyclopedia of Carbohydrate-Active Enzymes. Glycobiology 2018, 28, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Chernysheva, N.; Bystritskaya, E.; Stenkova, A.; Golovkin, I.; Nedashkovskaya, O.; Isaeva, M. Comparative Genomics and CAZyme Genome Repertoires of Marine Zobellia Amurskyensis KMM 3526T and Zobellia Laminariae KMM 3676T. Mar. Drugs 2019, 17, 661. [Google Scholar] [CrossRef] [PubMed]
- Leis, B.; Held, C.; Bergkemper, F.; Dennemarck, K.; Steinbauer, R.; Reiter, A.; Mechelke, M.; Moerch, M.; Graubner, S.; Liebl, W.; et al. Comparative Characterization of All Cellulosomal Cellulases from Clostridium Thermocellum Reveals High Diversity in Endoglucanase Product Formation Essential for Complex Activity. Biotechnol. Biofuels 2017, 10, 240. [Google Scholar] [CrossRef] [PubMed]
- Artzi, L.; Bayer, E.A.; Moraïs, S. Cellulosomes: Bacterial Nanomachines for Dismantling Plant Polysaccharides. Nat. Rev. Microbiol. 2017, 15, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Naas, A.E.; Mackenzie, A.K.; Mravec, J.; Schückel, J.; Willats, W.G.T.; Eijsink, V.G.H.; Pope, P.B. Do Rumen Bacteroidetes Utilize an Alternative Mechanism for Cellulose Degradation? mBio 2014, 5, e01401-14. [Google Scholar] [CrossRef] [PubMed]
- Grondin, J.M.; Tamura, K.; Déjean, G.; Abbott, D.W.; Brumer, H. Polysaccharide Utilization Loci: Fueling Microbial Communities. J. Bacteriol. 2017, 199, e00860-16. [Google Scholar] [CrossRef] [PubMed]
- Nakjang, S.; Ndeh, D.A.; Wipat, A.; Bolam, D.N.; Hirt, R.P. A Novel Extracellular Metallopeptidase Domain Shared by Animal Host-Associated Mutualistic and Pathogenic Microbes. PLoS ONE 2012, 7, e30287. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J. Families of Serine Peptidases. Methods Enzym. 1994, 244, 19–61. [Google Scholar]
- Accetto, T.; Avguštin, G. Polysaccharide Utilization Locus and CAZYme Genome Repertoires Reveal Diverse Ecological Adaptation of Prevotella Species. Syst. Appl. Microbiol. 2015, 38, 453–461. [Google Scholar] [CrossRef]
- Terrapon, N.; Lombard, V.; Drula, É.; Lapébie, P.; Al-Masaudi, S.; Gilbert, H.J.; Henrissat, B. PULDB: The Expanded Database of Polysaccharide Utilization Loci. Nucleic Acids Res. 2018, 46, D677–D683. [Google Scholar] [CrossRef]
- Yoshida, Y.; Sato, M.; Nonaka, T.; Hasegawa, Y.; Kezuka, Y. Characterization of the Phosphotransacetylase-Acetate Kinase Pathway for ATP Production in Porphyromonas Gingivalis. J. Oral Microbiol. 2019, 11, 1588086. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, E.L.; Xia, Q.; Wang, T.; Lamont, R.J.; Hackett, M. Pathway Analysis for Intracellular Porphyromonas Gingivalis Using a Strain ATCC 33277 Specific Database. BMC Microbiol. 2009, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Sato, T.; Yamada, T. Metabolic Pathways for Cytotoxic End Product Formation from Glutamate- and Aspartate-Containing Peptides by Porphyromonas Gingivalis. J. Bacteriol. 2000, 182, 4704–4710. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Sato, T. Preferential Utilization of Dipeptides by Porphyromonas Gingivalis. J. Dent. Res. 2001, 80, 1425–1429. [Google Scholar] [CrossRef]
- Spaans, S.K.; Weusthuis, R.A.; van der Oost, J.; Kengen, S.W.M. NADPH-Generating Systems in Bacteria and Archaea. Front. Microbiol 2015, 6, 742. [Google Scholar] [CrossRef]
- Kpebe, A.; Benvenuti, M.; Guendon, C.; Rebai, A.; Fernandez, V.; Le Laz, S.; Etienne, E.; Guigliarelli, B.; García-Molina, G.; de Lacey, A.L.; et al. A New Mechanistic Model for an O2-Protected Electron-Bifurcating Hydrogenase, Hnd from Desulfovibrio Fructosovorans. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 1302–1312. [Google Scholar] [CrossRef]
- Brandt, C.; Bongcam-Rudloff, E.; Müller, B. Abundance Tracking by Long-Read Nanopore Sequencing of Complex Microbial Communities in Samples from 20 Different Biogas/Wastewater Plants. Appl. Sci. 2020, 10, 7518. [Google Scholar] [CrossRef]
- Senés-Guerrero, C.; Colón-Contreras, F.A.; Reynoso-Lobo, J.F.; Tinoco-Pérez, B.; Siller-Cepeda, J.H.; Pacheco, A. Biogas-Producing Microbial Composition of an Anaerobic Digester and Associated Bovine Residues. Microbiol. Open 2019, 8, e00854. [Google Scholar] [CrossRef]
- Grohmann, A.; Fehrmann, S.; Vainshtein, Y.; Haag, N.L.; Wiese, F.; Stevens, P.; Naegele, H.-J.; Oechsner, H.; Hartsch, T.; Sohn, K.; et al. Microbiome Dynamics and Adaptation of Expression Signatures during Methane Production Failure and Process Recovery. Bioresour. Technol. 2018, 247, 347–356. [Google Scholar] [CrossRef]
- Bremer, E.; Krämer, R. Responses of Microorganisms to Osmotic Stress. Annu. Rev. Microbiol. 2019, 73, 313–334. [Google Scholar] [CrossRef]
- Booth, I.R.; Blount, P. The MscS and MscL Families of Mechanosensitive Channels Act as Microbial Emergency Release Valves. J. Bacteriol. 2012, 194, 4802–4809. [Google Scholar] [CrossRef] [PubMed]
- Heyer, R.; Kohrs, F.; Reichl, U.; Benndorf, D. Metaproteomics of Complex Microbial Communities in Biogas Plants. Microb. Biotechnol. 2015, 8, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, M. Metaproteomics: Much More than Measuring Gene Expression in Microbial Communities. mSystems 2019, 4, e00115-19. [Google Scholar] [CrossRef] [PubMed]
- Chiapello, M.; Zampieri, E.; Mello, A. A Small Effort for Researchers, a Big Gain for Soil Metaproteomics. Front. Microbiol. 2020, 11, 88. [Google Scholar] [CrossRef]
- Muth, T.; Behne, A.; Heyer, R.; Kohrs, F.; Benndorf, D.; Hoffmann, M.; Lehtevä, M.; Reichl, U.; Martens, L.; Rapp, E. The MetaProteomeAnalyzer: A Powerful Open-Source Software Suite for Metaproteomics Data Analysis and Interpretation. J. Proteome Res. 2015, 14, 1557–1565. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Chromosome |
|---|---|
| Genome size (bp) | 3,717,632 |
| GC content (%) | 47.97 |
| Total genes | 3055 |
| Protein coding genes | 3000 |
| rrn operons | 2 |
| tRNA genes | 49 |
| Locus Tag | EC No. | Enzyme | Reaction | Gene(s) |
|---|---|---|---|---|
| Catabolism of Glutamine and Glutamate | ||||
| ING2E5A_1298 | 3.5.1.2 | Glutaminase | l-glutamine + H2O <=> l-glutamate + NH3 | glsA |
| ING2E5A_2384 | 1.4.1.13 | Glutamate synthase (NADPH) | 2 l-glutamate + NADP( + ) <=> l-glutamine + 2-oxoglutarate + NADPH | gltD |
| ING2E5A_0283, ING2E5A_2397 | 1.4.1.3, 1.4.1.2 | Glutamate dehydrogenase (NAD(P) +), Glutamate dehydrogenase | l-glutamate + H2O + NAD(P)+ <=> 2-oxoglutarate + NH3 + NAD(P)H, l-glutamate + H2O + NAD+ <=> 2-oxoglutarate + NH3 + NADH | gdhA gdh |
| ING2E5A_2385 | 1.4.7.1 | Ferredoxin-dependent glutamate synthase 1 | 2 l-glutamate + 2 oxidized ferredoxin <=> l-glutamine + 2-oxoglutarate + 2 reduced ferredoxin + 2 H+ | gltB |
| ING2E5A_1056, ING2E5A_1057 | 1.2.7.3 | 2-oxoglutarate synthase | 2-oxoglutarate + CoA + 2 oxidized ferredoxin <=> succinyl-CoA + CO2 + 2 reduced ferredoxin + 2 H+ | korB, korA |
| ING2E5A_1055, ING2E5A_1054 | 1.2.4.2, 2.3.1.61 | Oxoglutarate dehydrogenase (succinyl-transferring), Dihydrolipoyllysine-residue succinyltransferase | 2-oxoglutarate → succinyl-CoA | sucA, sucB |
| ING2E5A_1515, ING2E5A_1517 | 5.4.99.2 | Methylmalonyl-CoA mutase | (R)-methylmalonyl-CoA <=> succinyl-CoA | mutA, mutB |
| ING2E5A_1046 | 5.1.99.1 | Methylmalonyl-CoA epimerase | (R)-methylmalonyl-CoA <=> (S)-2-Methylmalonyl-CoA | |
| ING2E5A_1045, ING2E5A_1864 | 6.4.1.3 | Propionyl-CoA carboxylase | ATP + propanoyl-CoA + HCO3- <=> ADP + phosphate + (S)-methylmalonyl-CoA | pccB1 pccB3 |
| ING2E5A_0730 | 2.3.1.8 | Phosphate acetyltransferase | Acetyl-CoA + phosphate <=> CoA + acetyl phosphate | pta |
| ING2E5A_0728 | 2.7.2.1 | Acetate kinase | ATP + acetate <=> ADP + acetyl phosphate | ackA |
| Catabolism of Arginine and Aspartate | ||||
| ING2E5A_2671 | 3.5.1.1 | l-asparaginase 1 | l-asparagine + H2O <=> l-aspartate + NH3 | ansA |
| ING2E5A_2287 | 4.3.1.1 | Aspartate ammonia-lyase | l-aspartate <=> fumarate + NH3 | aspA |
| ING2E5A_3067 | 1.4.3.16 | l-aspartate oxidase | l-aspartate + O2 <=> iminosuccinate + H2O2 | nadB |
| ING2E5A_1308 | 2.6.1.1 | Aspartate transaminase | l-aspartate + 2-oxoglutarate <=> oxaloacetate + l-glutamate | aspC |
| ING2E5A_2589 | 3.5.3.6 | Arginine deiminase | l-arginine + H2O <=> l-citrulline + NH3 | arcA |
| ING2E5A_0737 | 6.3.4.5 | Argininosuccinate synthase | ATP + l-citrulline + l-aspartate <=> AMP + diphosphate + N(omega)-(l-arginino)succinate | argG |
| ING2E5A_0731 | 4.3.2.1 | Argininosuccinate lyase | 2-(N(omega)-l-arginino)succinate <=> fumarate + l-arginine | argH |
| ING2E5A_3031, ING2E5A_3032, ING2E5A_3033 | 1.3.5.1 | Succinate dehydrogenase, Succinate dehydrogenase flavoprotein subunit | Succinate + a quinone <=> fumarate + a quinol | sdhA |
| ING2E5A_0952, ING2E5A_0953 | 6.2.1.5 | Succinate-CoA ligase (ADP-forming) | ATP + succinate + CoA <=> ADP + phosphate + succinyl-CoA | sucD, sucC |
| ING2E5A_0662 | 4.2.1.2 | Fumarate hydratase | (S)-malate <=> fumarate + H2O | fumA |
| ING2E5A_2725 | 1.1.1.37 | Malate dehydrogenase | (S)-malate + NAD+ <=> oxaloacetate + NADH | mdh |
| ING2E5A_1041, ING2E5A_1042 | 4.1.1.112 | Oxaloacetate decarboxylase | Oxaloacetate <=> pyruvate + CO2 | oadB2-1, oadB2-2 |
| ING2E5A_1825 | 4.1.1.112 | Oxaloacetate decarboxylase | Oxaloacetate <=> pyruvate + CO2 | oadB |
| ING2E5A_0270, ING2E5A_1826 | 6.4.1.1 | Pyruvate carboxylase | ATP + pyruvate + HCO3- <=> ADP + phosphate + oxaloacetate | pycB1, pycB3 |
| ING2E5A_1490, ING2E5A_1805, ING2E5A_1806, | 1.2.4.1 | Pyruvate dehydrogenase | Pyruvate + [dihydrolipoyllysine-residue acetyltransferase] lipoyllysine <=> [dihydrolipoyllysine-residue acetyltransferase] S-acetyldihydrolipoyllysine + CO(2) | pdhB1, pdhB3, pdhA |
| ING2E5A_1158, ING2E5A_1264, ING2E5A_1491, ING2E5A_1804 | 2.3.1.12 | Dihydrolipoyllysine-residue acetyltransferase | Acetyl-CoA + enzyme N(6)-(dihydrolipoyl)lysine <=> CoA + enzyme N(6)-(S-acetyldihydrolipoyl)lysine | pdhC1, dlaT, pdhC3, pdhC5 |
| ING2E5A_0730 | 2.3.1.8 | Phosphate acetyltransferase | Acetyl-CoA + phosphate <=> CoA + acetyl phosphate | pta |
| ING2E5A_0728 | 2.7.2.1 | Acetate kinase | ATP + acetate <=> ADP + acetyl phosphate | ackA |
| Butanoate pathway enzymes | ||||
| ING2E5A_0177 | 2.7.2.7 | Butyrate kinase | ATP + butanoate <=> ADP + butanoyl phosphate | buk |
| ING2E5A_0176 | 2.3.1.19 | Phosphate butyryltransferase | Butanoyl-CoA + phosphate <=> CoA + butanoylphosphate | ptb |
| ING2E5A_0729 | 1.1.1.157 | 3-hydroxybutyryl-CoA dehydrogenase | (S)-3-hydroxybutanoyl-CoA + NADP+ <=> 3-acetoacetyl-CoA + NADPH | hbd |
| Malic Enzyme Module | ||||
| ING2E5A_2598 | 1.1.1.40 | Malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+). | (S)-malate + NADP+ <=> pyruvate + CO2 + NADPH, Oxaloacetate <=> pyruvate + CO2 | maeB |
| ING2E5A_3022 | 2.7.9.1 | Pyruvate, phosphate dikinase | ATP + pyruvate + phosphate <=> AMP + phosphoenolpyruvate + diphosphate | ppdK |
| Ranking | GenID | Gene Length | Putative Gene Product | Gene | EC Number | Transcriptome Reads Mapped | Normalized Number of Transcripts (TPM) |
|---|---|---|---|---|---|---|---|
| 7 | ING2E5A_1308 | 1191 | Aspartate aminotransferase | ast | 2.6.1.1 | 739 | 21,591 |
| 8 | ING2E5A_1484 | 1179 | 6-phosphogluconolactonase | pgls | 3.1.1.31 | 178 | 5255 |
| 21 | ING2E5A_1407 | 1500 | Propionyl-CoA:succinate CoA transferase | scpC | 2.8.3.- | 186 | 4328 |
| 26 | ING2E5A_0536 | 720 | Peptidase E | pepE | 3.4.13.21 | 76 | 3688 |
| 32 | ING2E5A_3029 | 1191 | Aspartate aminotransferase | ast | 2.6.1.1 | 113 | 3315 |
| 37 | ING2E5A_0388 | 657 | Uracil phosphoribosyltransferase | uprt | 2.4.2.9 | 57 | 3049 |
| 43 | ING2E5A_2638 | 1521 | Periplasmic serine endoprotease | degP | 3.4.21.107 | 121 | 2774 |
| 50 | ING2E5A_0405 | 1452 | 6-phosphogluconate dehydrogenase, decarboxylating | gndA | 1.1.1.44 | 97 | 2344 |
| 85 | ING2E5A_0914 | 3792 | TonB-dependent outer-membrane transporter | susC | n.d. | 205 | 1887 |
| 94 | ING2E5A_0692 | 1014 | ATP-dependent 6-phosphofructokinase | pfk | 2.7.1.11 | 50 | 1725 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maus, I.; Tubbesing, T.; Wibberg, D.; Heyer, R.; Hassa, J.; Tomazetto, G.; Huang, L.; Bunk, B.; Spröer, C.; Benndorf, D.; et al. The Role of Petrimonas mucosa ING2-E5AT in Mesophilic Biogas Reactor Systems as Deduced from Multiomics Analyses. Microorganisms 2020, 8, 2024. https://doi.org/10.3390/microorganisms8122024
Maus I, Tubbesing T, Wibberg D, Heyer R, Hassa J, Tomazetto G, Huang L, Bunk B, Spröer C, Benndorf D, et al. The Role of Petrimonas mucosa ING2-E5AT in Mesophilic Biogas Reactor Systems as Deduced from Multiomics Analyses. Microorganisms. 2020; 8(12):2024. https://doi.org/10.3390/microorganisms8122024
Chicago/Turabian StyleMaus, Irena, Tom Tubbesing, Daniel Wibberg, Robert Heyer, Julia Hassa, Geizecler Tomazetto, Liren Huang, Boyke Bunk, Cathrin Spröer, Dirk Benndorf, and et al. 2020. "The Role of Petrimonas mucosa ING2-E5AT in Mesophilic Biogas Reactor Systems as Deduced from Multiomics Analyses" Microorganisms 8, no. 12: 2024. https://doi.org/10.3390/microorganisms8122024
APA StyleMaus, I., Tubbesing, T., Wibberg, D., Heyer, R., Hassa, J., Tomazetto, G., Huang, L., Bunk, B., Spröer, C., Benndorf, D., Zverlov, V., Pühler, A., Klocke, M., Sczyrba, A., & Schlüter, A. (2020). The Role of Petrimonas mucosa ING2-E5AT in Mesophilic Biogas Reactor Systems as Deduced from Multiomics Analyses. Microorganisms, 8(12), 2024. https://doi.org/10.3390/microorganisms8122024