Comparative Metagenomics of Palearctic and Neotropical Avian Cloacal Viromes Reveal Geographic Bias in Virus Discovery

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Selection
2.3. Sample Processing and Next Generation Sequencing
2.4. Detection of Individuals Positive for the Viruses
2.5. Genomic Analysis of the Novel Viruses
2.6. Phylogenetic and Taxonomic Analysis
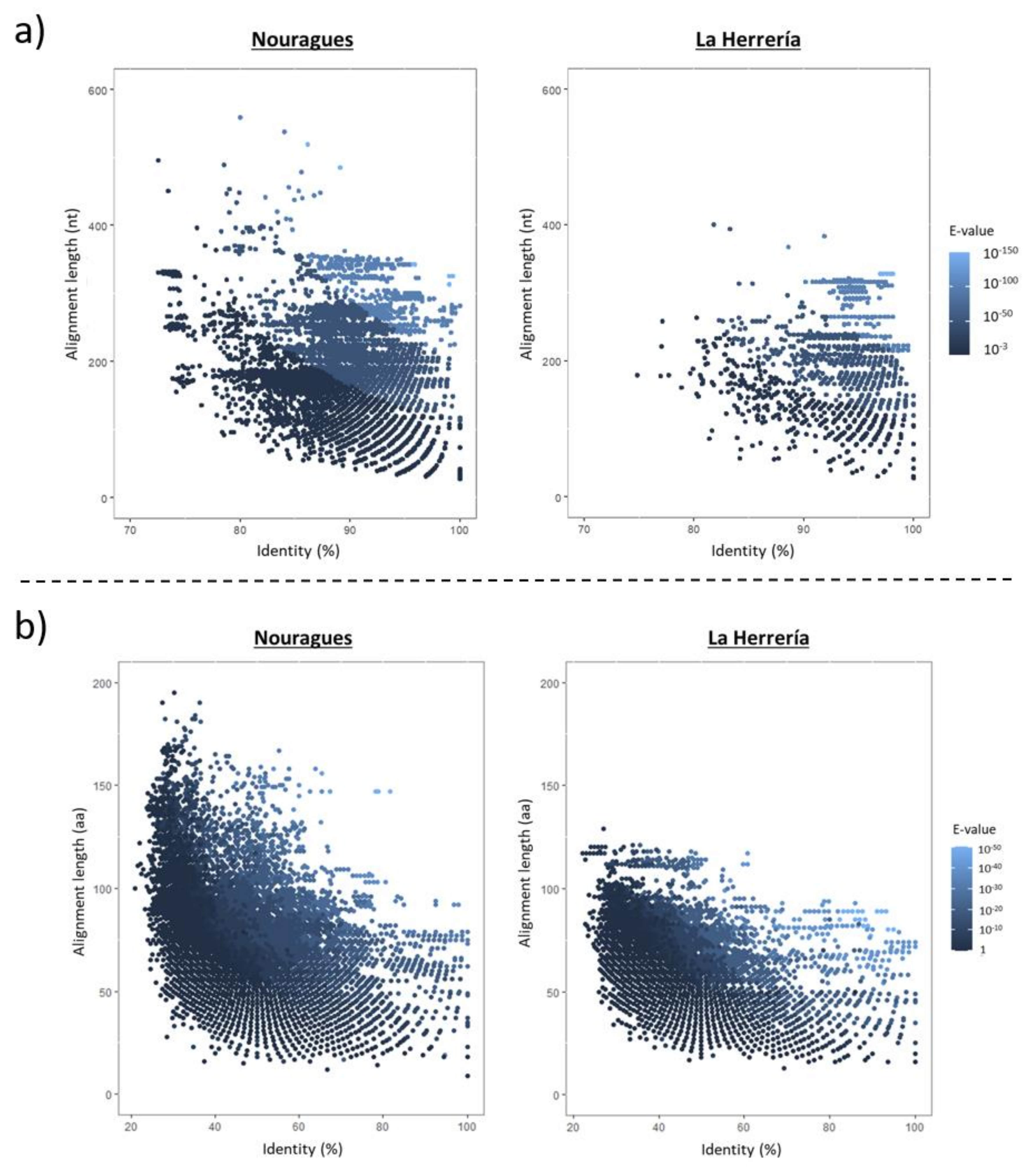
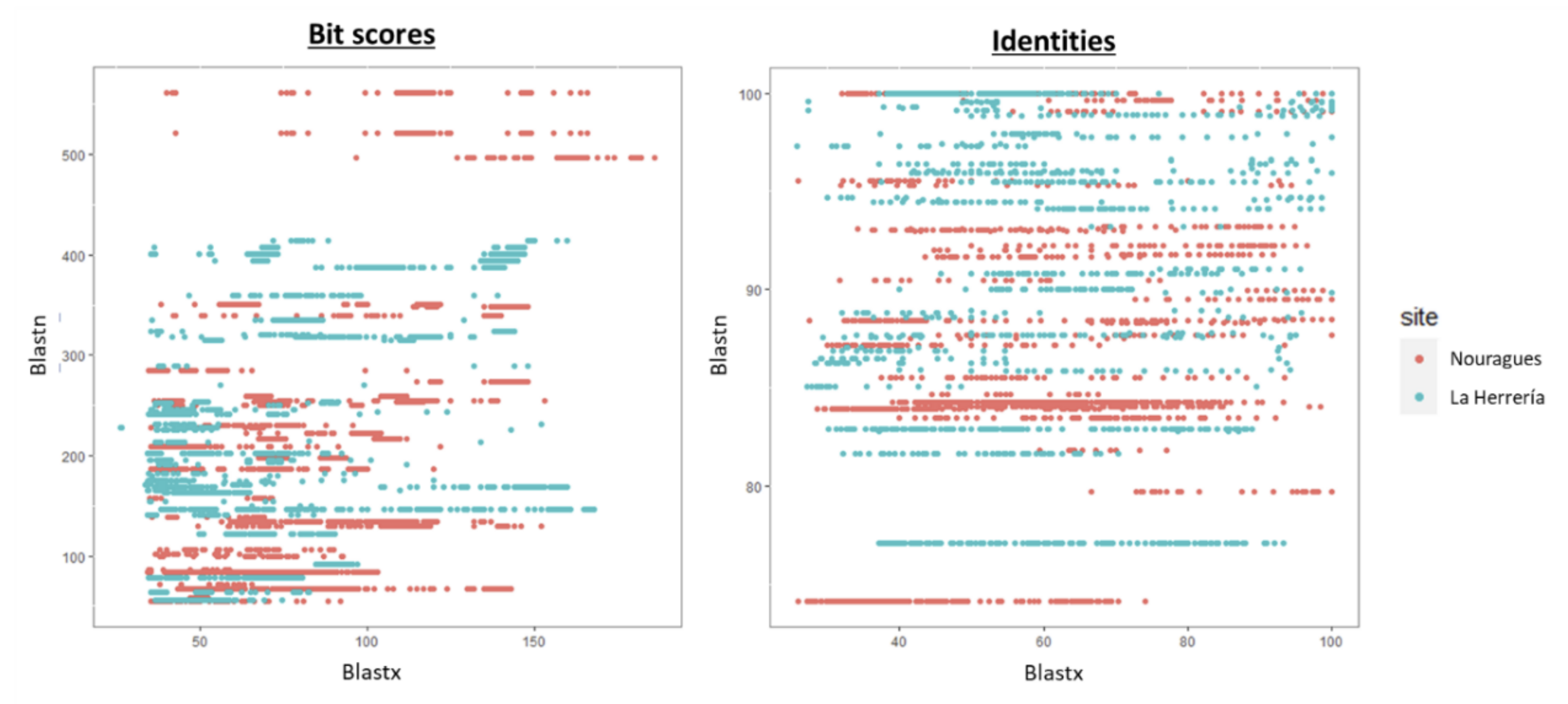
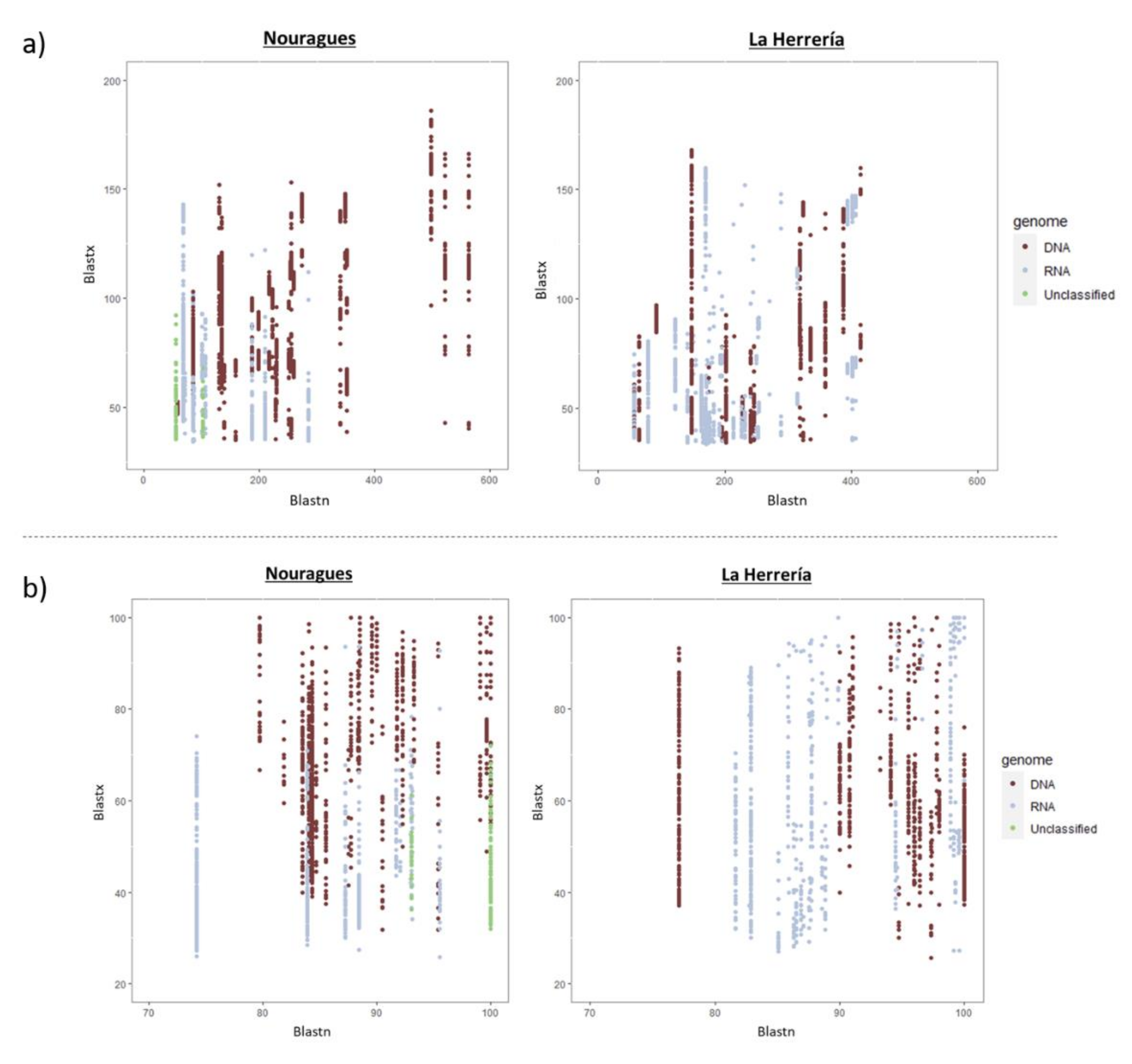
2.7. Comparative Analysis
3. Results
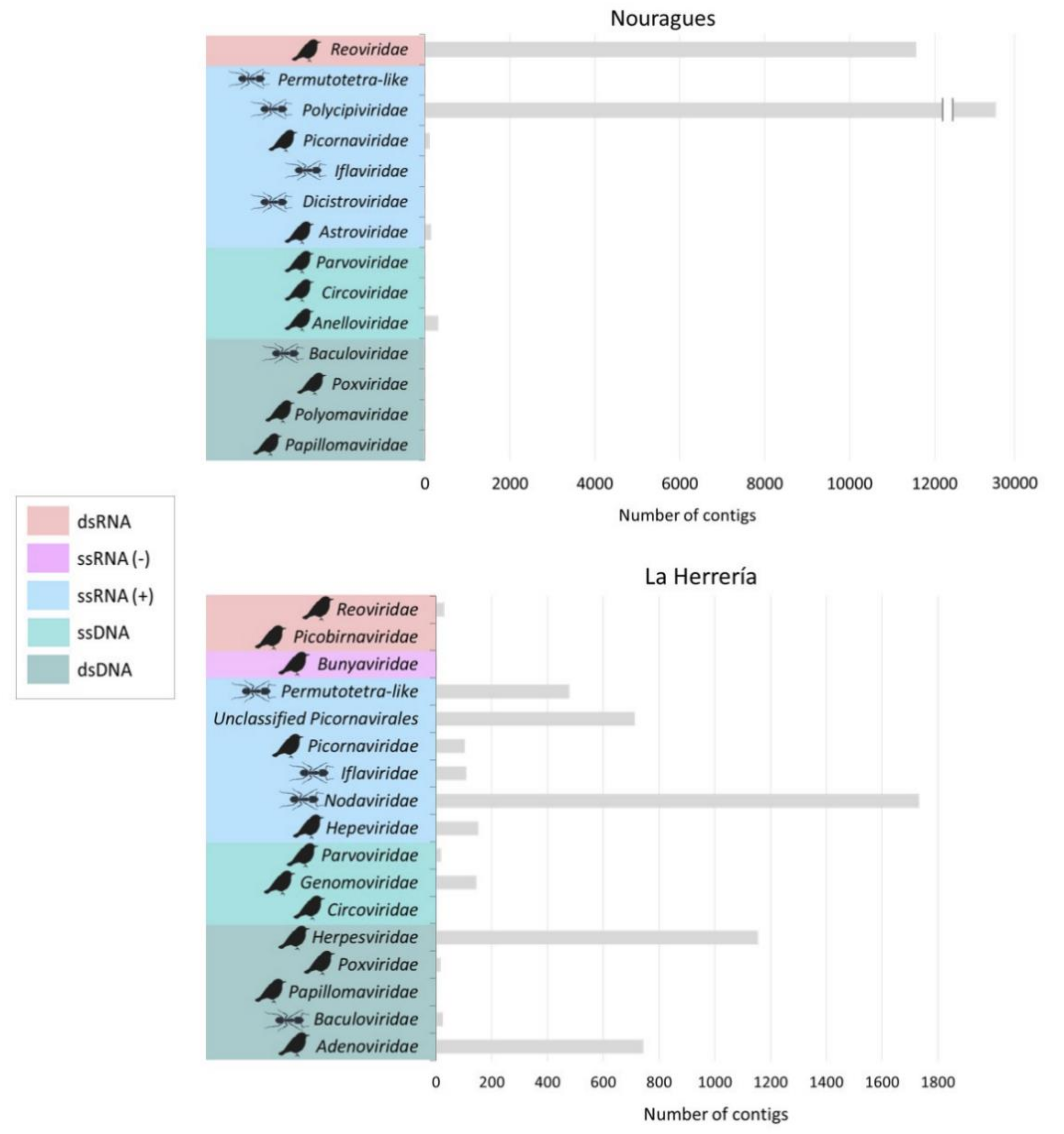
3.1. Composition of Nouragues/La Herrería Cloacal Viromes
3.2. Novel Viruses Found in Nouragues/La Herrería
4. Discussion
4.1. Composition of Nouragues/La Herrería Cloacal Viromes
4.2. Novel Viruses of Interest Found in Nouragues/La Herrería
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
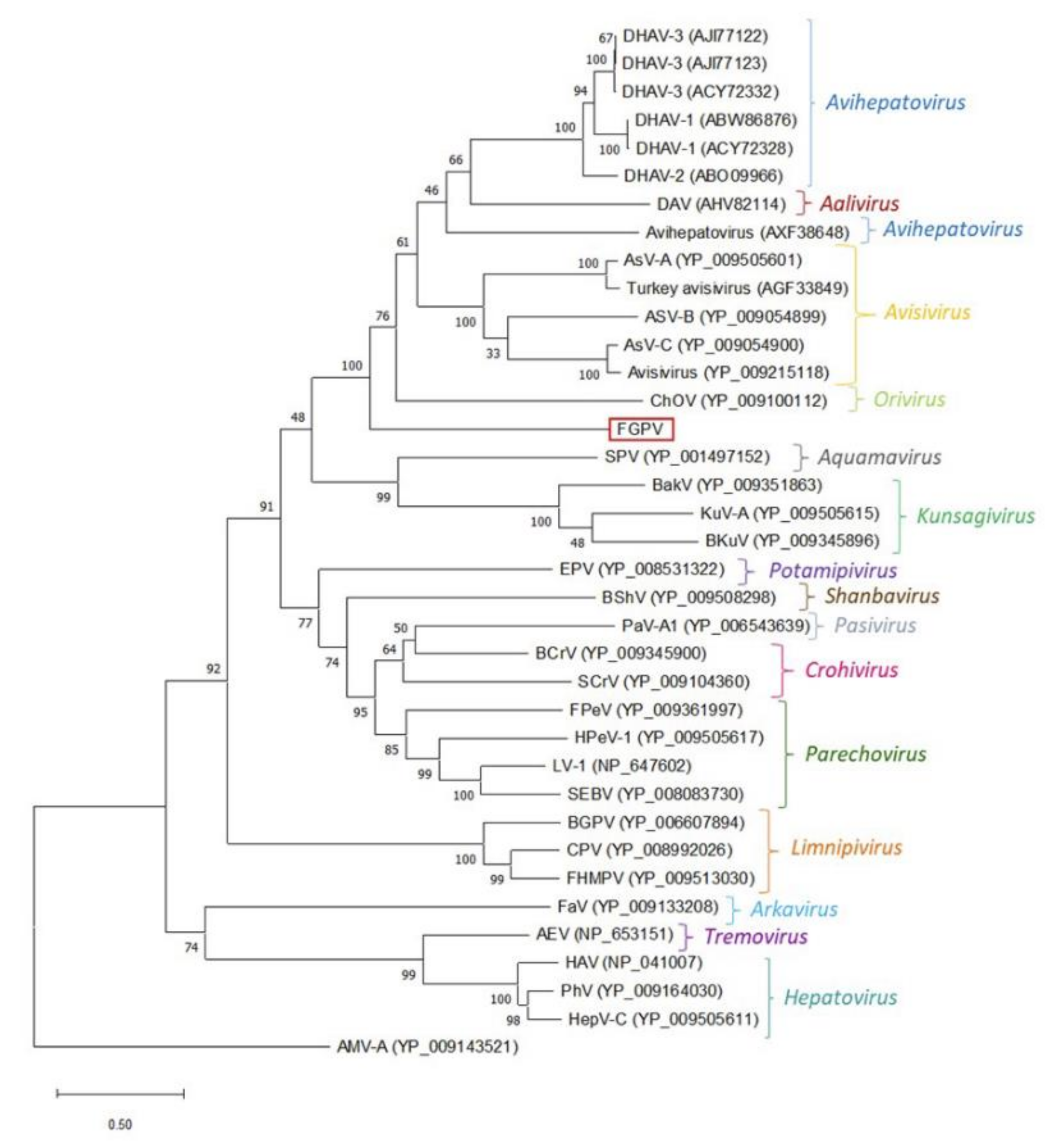
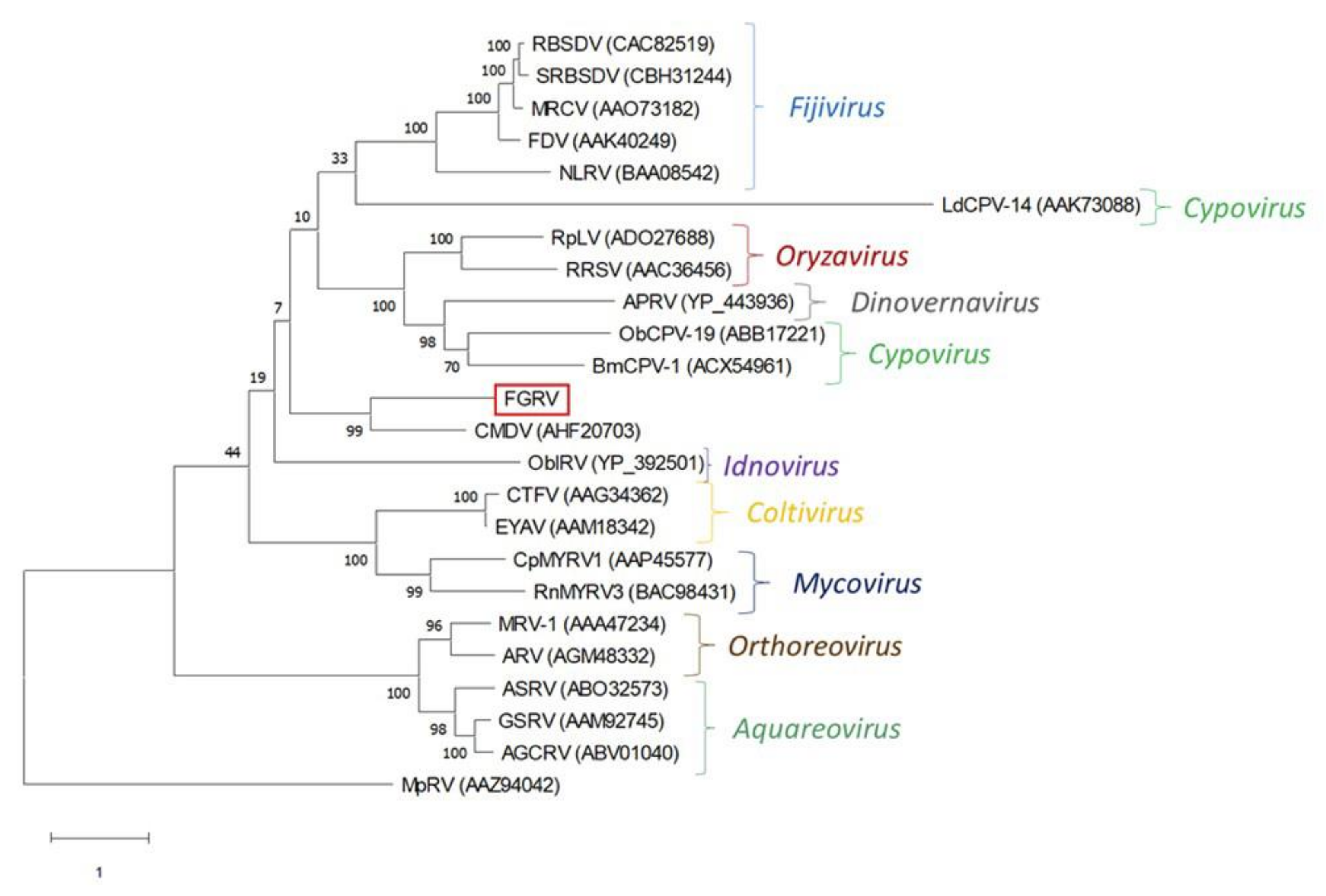
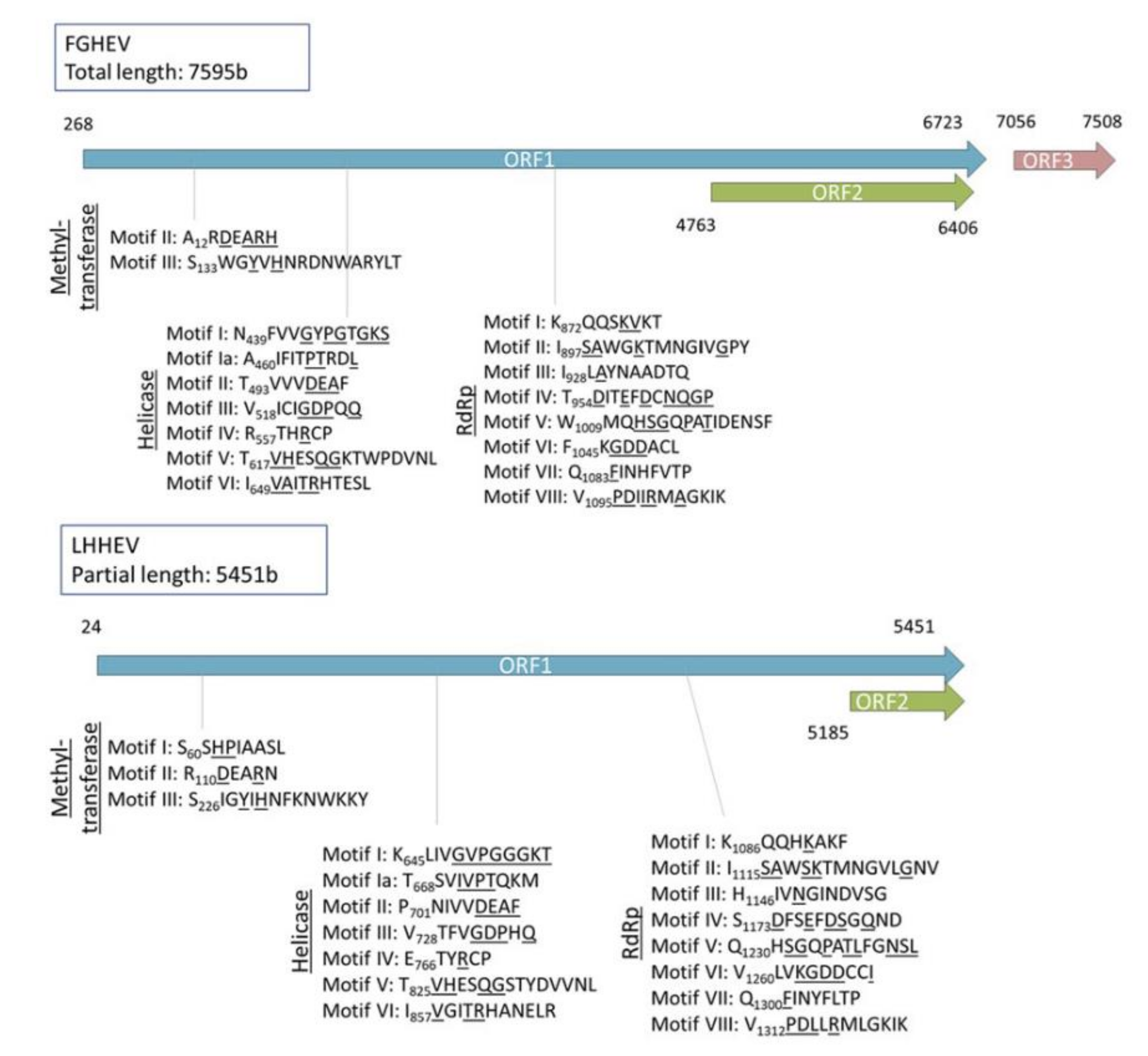
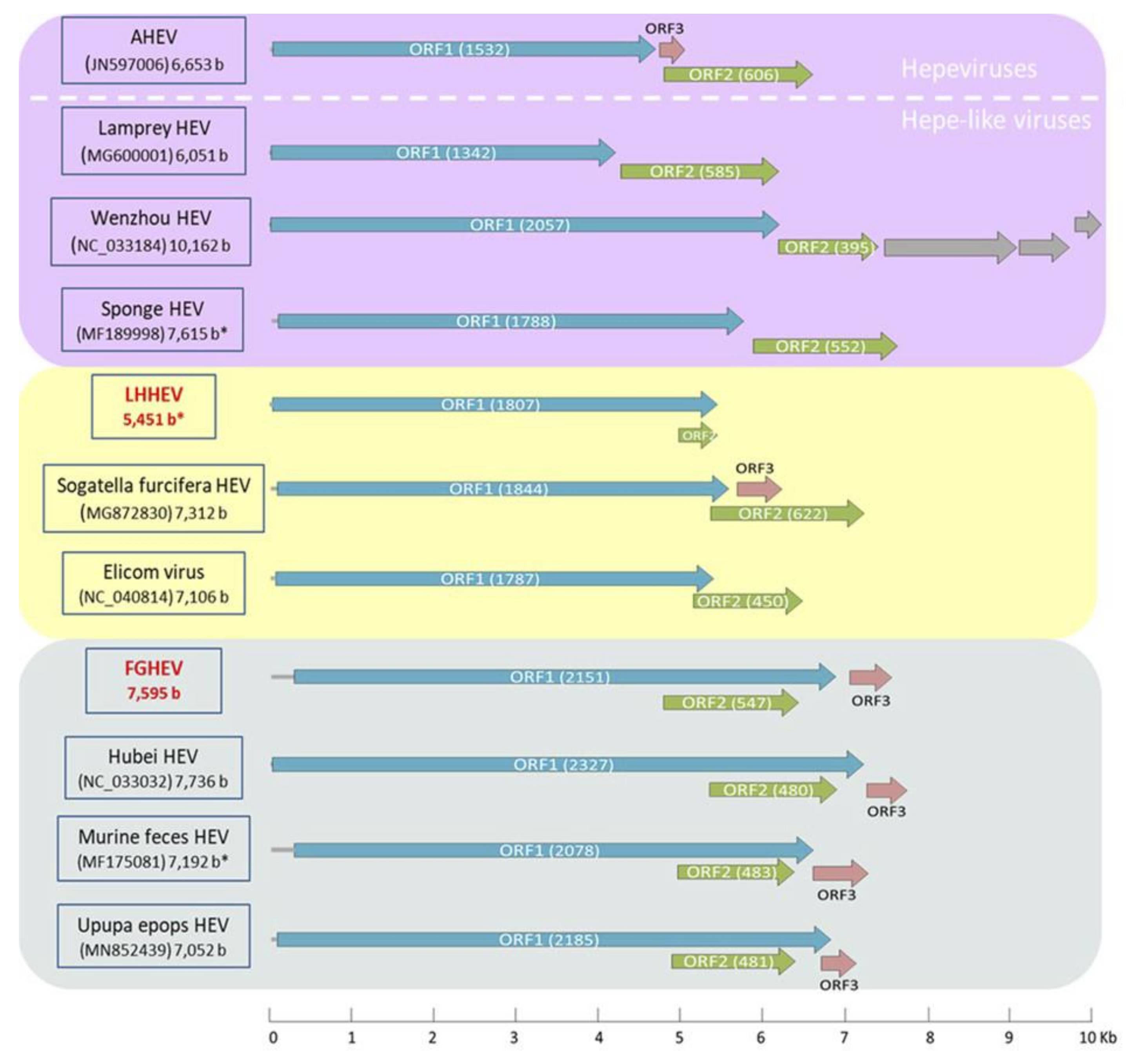
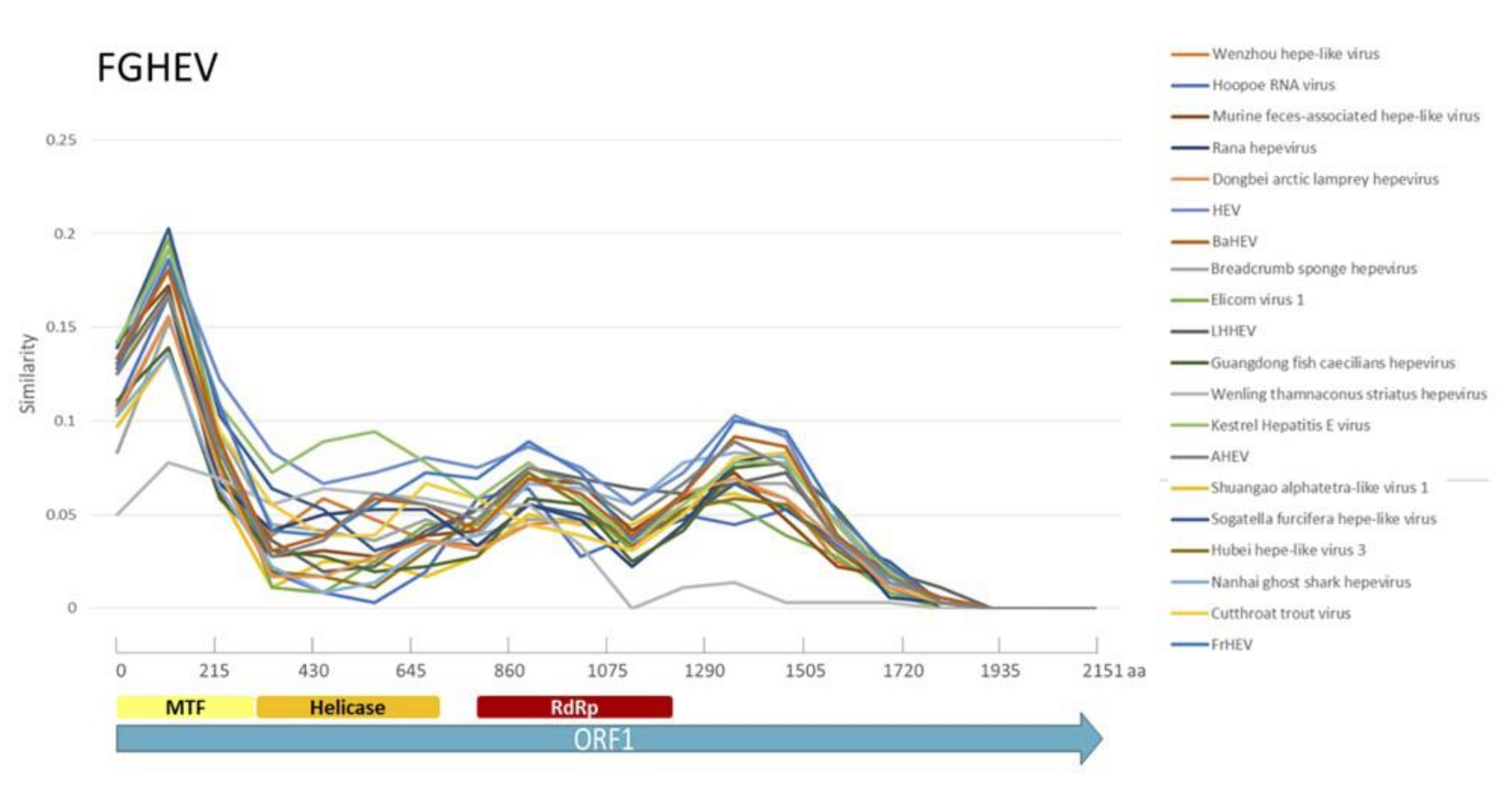
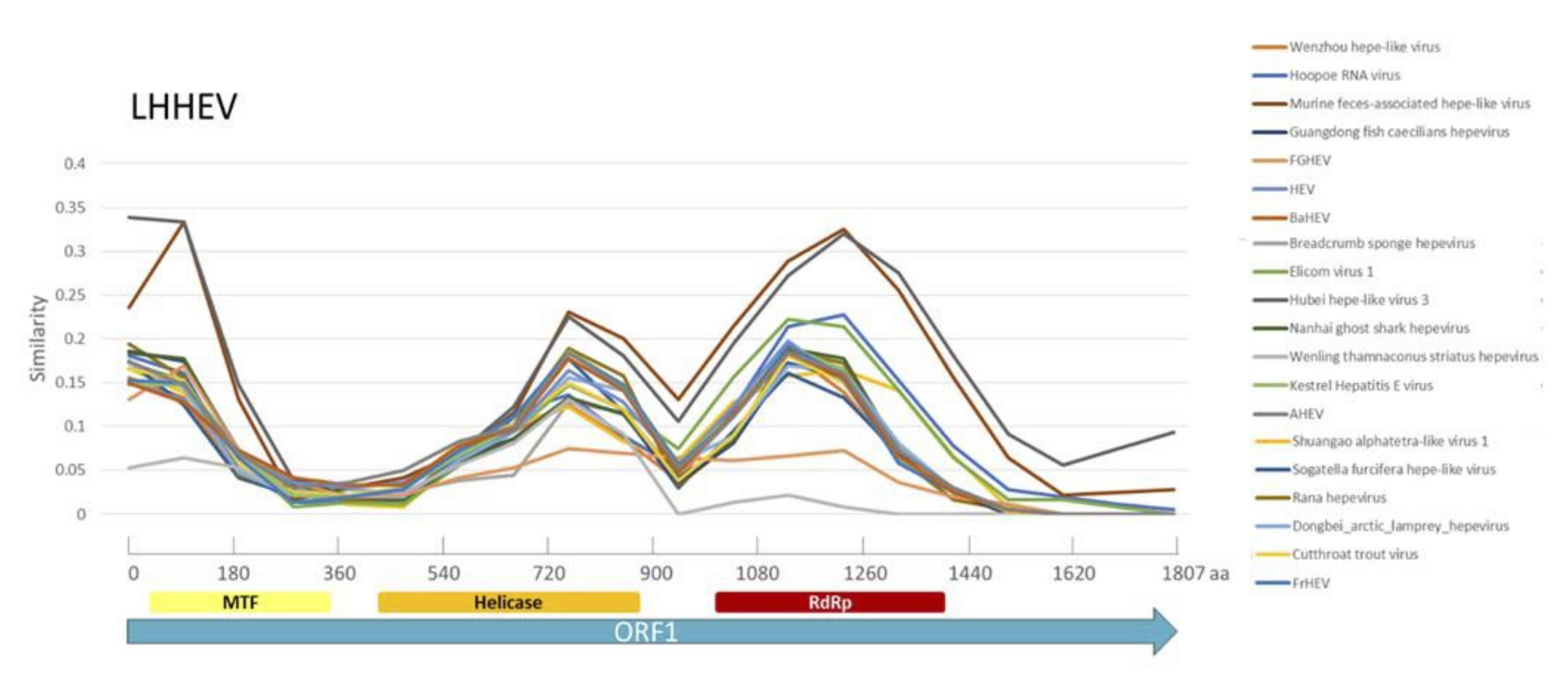
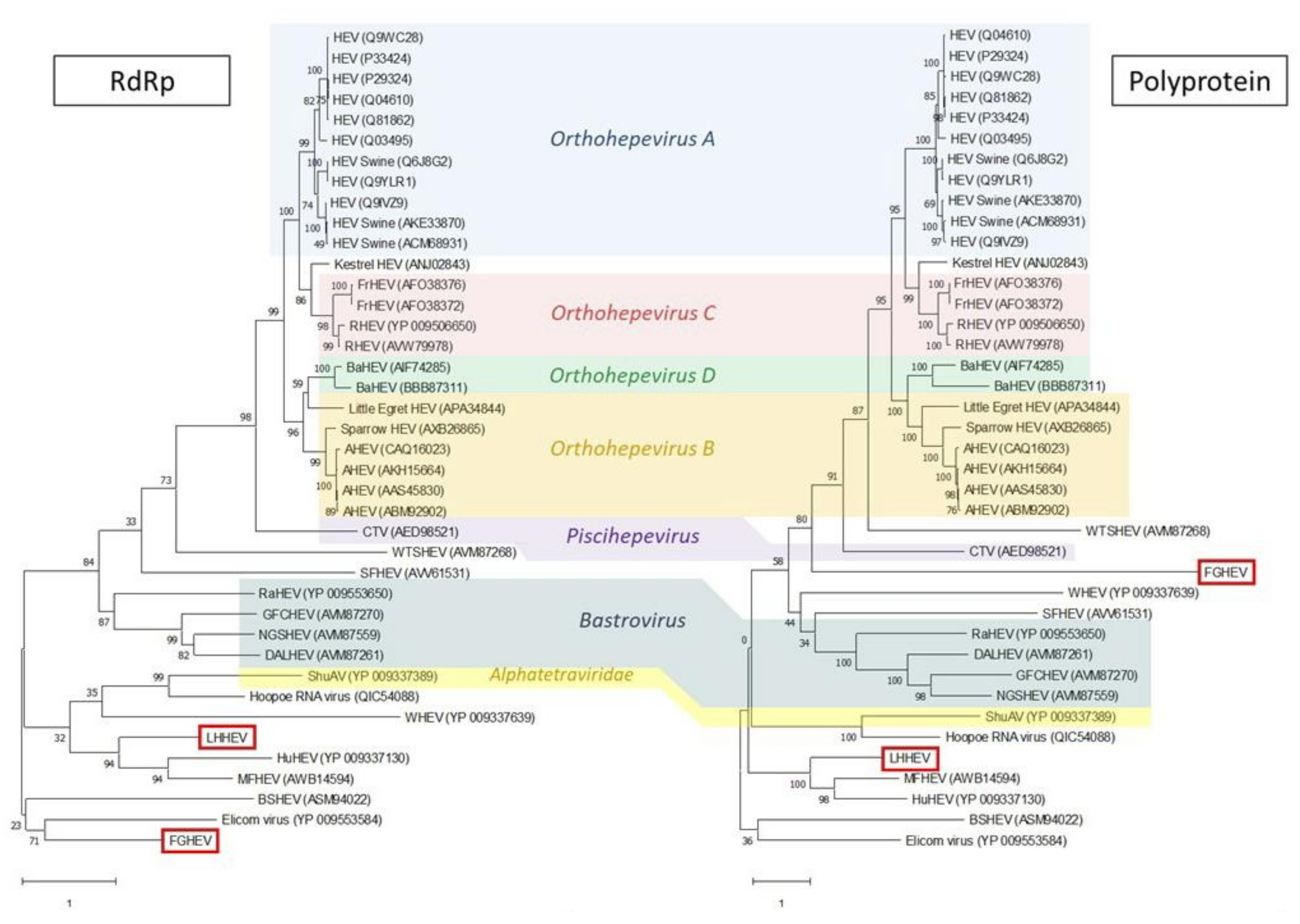
Appendix A. Description of the Novel Viruses of Interest Found in Nouragues and La Herrería







References
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Méndez, A.; Tomori, O.; Mazet, J.A.K. The Global Virome Project. Science 2018, 359, 872–874. [Google Scholar] [CrossRef] [PubMed]
- el Zowalaty, M.E.; Järhult, J.D. From SARS to COVID-19: A previously unknown SARS- related coronavirus (SARS-CoV-2) of pandemic potential infecting humans—Call for a One Health approach. One Health 2020, 9, 100124. [Google Scholar] [CrossRef] [PubMed]
- Lebov, J.; Grieger, K.; Womack, D.; Zaccaro, D.; Whitehead, N.; Kowalcyk, B.; MacDonald, P.D.M. A framework for One Health research. One Health 2017, 3, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Bazán, E.R. Symbiosis: Viruses as intimate partners. Annu. Rev. Virol. 2017, 4, 123–139. [Google Scholar] [CrossRef]
- Barrowclough, G.F.; Cracraft, J.; Klicka, J.; Zink, R.M. How many kinds of birds are there and why does it matter? PLoS ONE 2016, 11, e0166307. [Google Scholar] [CrossRef]
- Benskin, C.M.W.H.; Wilson, K.; Jones, K.; Hartley, I.R. Bacterial pathogens in wild birds: A review of the frequency and effects of infection. Biol. Rev. 2009, 84, 349–373. [Google Scholar] [CrossRef]
- Dhama, K.; Mahendran, M.; Tomar, S. Pathogens transmitted by migratory birds: Threat perceptions to poultry health and production. Int. J. Poult. Sci. 2008, 7, 516–525. [Google Scholar] [CrossRef]
- Viana, D.S.; Santamaría, L.; Figuerola, J. Migratory birds as global dispersal vectors. Trends Ecol. Evol. 2016, 31, 763–775. [Google Scholar] [CrossRef]
- Hulse-Post, D.J.; Sturm-Ramirez, K.M.; Humberd, J.; Seiler, P.; Govorkova, E.A.; Krauss, S.; Scholtissek, C.; Puthavathana, P.; Buranathai, C.; Nguyen, T.D.; et al. Role of domestic ducks in the propagation and biological evolution of highly pathogenic H5N1 influenza viruses in Asia. Proc. Natl. Acad. Sci. USA 2005, 102, 10682–10687. [Google Scholar] [CrossRef]
- Wille, M.; Eden, J.S.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virus–virus interactions and host ecology are associated with RNA virome structure in wild birds. Mol. Ecol. 2018, 27, 5263–5278. [Google Scholar] [CrossRef]
- Chan, J.F.W.; To, K.K.W.; Chen, H.; Yuen, K.Y. Cross-species transmission and emergence of novel viruses from birds. Curr. Opin. Virol. 2015, 10, 63–69. [Google Scholar] [CrossRef] [PubMed]
- François, S.; Pybus, O.G. Towards an understanding of the avian virome. J. Gen. Virol. 2020, jgv001447. [Google Scholar] [CrossRef] [PubMed]
- Michel, F.; Fischer, D.; Eiden, M.; Fast, C.; Reuschel, M.; Müller, K.; Rinder, M.; Urbaniak, S.; Brandes, F.; Schwehn, R.; et al. West Nile virus and Usutu virus monitoring of wild birds in Germany. Int. J. Environ. Res. Public Health 2018, 15, 171. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.R.; Bevins, S.N. A review of virulent Newcastle disease viruses in the United States and the role of wild birds in viral persistence and spread. Vet. Res. 2017, 48, 1–15. [Google Scholar] [CrossRef]
- Li, M.; Liu, H.; Bi, Y.; Sun, J.; Wong, G.; Liu, D.; Li, L.; Liu, J.; Chen, Q.; Wang, H.; et al. Highly pathogenic avian influenza A(H5N8) virus in wild migratory birds, Qinghai Lake, China. Emerg. Infect. Dis. 2017, 23, 637–641. [Google Scholar] [CrossRef]
- Fawaz, M.; Vijayakumar, P.; Mishra, A.; Gandhale, P.N.; Dutta, R.; Kamble, N.M.; Sudhakar, S.B.; Roychoudhary, P.; Kumar, H.; Kulkarni, D.D.; et al. Duck gut viral metagenome analysis captures snapshot of viral diversity. Gut Pathog. 2016, 8, 30. [Google Scholar] [CrossRef]
- Ramírez-Martínez, L.A.; Loza-Rubio, E.; Mosqueda, J.; González-Garay, M.L.; García-Espinosa, G. Fecal virome composition of migratory wild duck species. PLoS ONE 2018, 13, e0206970. [Google Scholar] [CrossRef]
- Zhao, L.; Niu, Y.; Lu, T.; Yin, H.; Zhang, Y.; Xu, L.; Wang, Y.; Chen, H. Metagenomic analysis of the Jinding duck fecal virome. Curr. Microbiol. 2018, 75, 658–665. [Google Scholar] [CrossRef]
- Cracraft, J.; Barker, F.K. Passerine birds (Passeriformes). In Timetree of Life; Hedges, S.B., Kumar, S., Eds.; Oxford University Press: New York, NY, USA, 2009; pp. 423–431. [Google Scholar]
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.L.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal virome analysis of wild animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef]
- Thiollay, J.M. Avian diversity and distribution in French Guiana: Patterns across a large forest landscape. J. Trop. Ecol. 2002, 471–498. [Google Scholar] [CrossRef]
- Bodewes, R. Novel viruses in birds: Flying through the roof or is a cage needed? Vet. J. 2018, 233, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.C.; Navarro Lopez, R.; Morales, R.; Olvera, M.A.; Marquez, M.A.; Merino, R.; Miller, P.J.; Afonsoa, C.L. Molecular epidemiology of newcastle disease in Mexico and the potential spillover of viruses from poultry into wild bird species. Appl. Environ. Microbiol. 2013, 79, 4985–4992. [Google Scholar] [CrossRef] [PubMed]
- Rohaim, M.A.; el Naggar, R.F.; Helal, A.M.; Hussein, H.A.; Munir, M. Reverse spillover of avian viral vaccine strains from domesticated poultry to wild birds. Vaccine 2017, 35, 3523–3527. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E. Animal virus discovery: Improving animal health, understanding zoonoses, and opportunities for vaccine development. Curr. Opin. Virol. 2012, 2, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, M.; Riera, B. Geography and Climate. In Monographiae Biologicae; Dumont, H.J., Ed.; Kluwer Academic: Berlin, Germeny, 2001; pp. 9–18. [Google Scholar]
- Font, I. Climatología de España y Portugal; Universidad de Salamanca: Salamanca, Spain, 2000. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition—Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Fernández-Correa, I.; Truchado, D.A.; Gomez-Lucia, E.; Doménech, A.; Pérez-Tris, J.; Schmidt-Chanasit, J.; Cadar, D.; Benítez, L. A novel group of avian astroviruses from Neotropical passerine birds broaden the diversity and host range of Astroviridae. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Truchado, D.A.; Diaz-Piqueras, J.M.; Gomez-Lucia, E.; Doménech, A.; Milá, B.; Pérez-Tris, J.; Schmidt-Chanasit, J.; Cadar, D.; Benítez, L. A novel and divergent gyrovirus with unusual genomic features detected in wild passerine birds from a remote rainforest in French Guiana. Viruses 2019, 11, 1148. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; ISBN 3-900051-07-0. [Google Scholar]
- Oksanen, J. Vegan: Ecological Diversity. Available online: http://mirror.linux.duke.edu/cran/web/packages/vegan/vignettes/diversity-vegan.pdf (accessed on 24 September 2020).
- Sun, J.; He, W.T.; Wang, L.; Lai, A.; Ji, X.; Zhai, X.; Li, G.; Suchard, M.A.; Tian, J.; Zhou, J.; et al. COVID-19: Epidemiology, evolution, and cross-disciplinary perspectives. Trends Mol. Med. 2020, 26, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Vibin, J.; Chamings, A.; Collier, F.; Klaassen, M.; Nelson, T.M.; Alexandersen, S. Metagenomics detection and characterisation of viruses in faecal samples from Australian wild birds. Sci. Rep. 2018, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, S.; Liu, D.; Zhou, C.; Li, W.; Lin, Y.; Wang, X.; Shen, Q.; Wang, H.; Li, C.; et al. The fecal virome of red-crowned cranes. Arch. Virol. 2019, 164, 3–16. [Google Scholar] [CrossRef]
- Lima, D.A.; Cibulski, S.P.; Finkler, F.; Teixeira, T.F.; Varela, A.P.M.; Cerva, C.; Loiko, M.R.; Scheffer, C.M.; Dos Santos, H.F.; Mayer, F.Q.; et al. Faecal virome of healthy chickens reveals a large diversity of the eukaryote viral community, including novel circular ssDNA viruses. J. Gen. Virol. 2017, 98, 690–703. [Google Scholar] [CrossRef]
- Swayne, D.E. Avian Influenza; Blackwell Pub: New York, NY, USA, 2008; ISBN 9780813820477. [Google Scholar]
- Webster, R.G.; Yakhno, M.; Hinshaw, V.S.; Bean, W.J.; Gopal Murti, K. Intestinal influenza: Replication and characterization of influenza viruses in ducks. Virology 1978, 84, 268–278. [Google Scholar] [CrossRef]
- Wille, M.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virome heterogeneity and connectivity in waterfowl and shorebird communities. ISME J. 2019, 13, 2603–2616. [Google Scholar] [CrossRef]
- Slusher, M.J.; Wilcox, B.R.; Lutrell, M.P.; Poulson, R.L.; Brown, J.D.; Yabsley, M.J.; Stallknecht, D.E. Are passerine birds reservoirs for influenza a viruses? J. Wildl. Dis. 2014, 50, 792–809. [Google Scholar] [CrossRef]
- Rosseel, T.; Ozhelvaci, O.; Freimanis, G.; van Borm, S. Evaluation of convenient pretreatment protocols for RNA virus metagenomics in serum and tissue samples. J. Virol. Methods 2015, 222, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Vo, N.P.; Boros, Á.; Pankovics, P.; Reuter, G.; Li, O.T.W.; Wang, C.; Deng, X.; Poon, L.L.M.; Delwart, E. The viruses of wild pigeon droppings. PLoS ONE 2013, 8, e72787. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef]
- Temmam, S.; Hul, V.; Bigot, T.; Hoem, T.; Gorman, C.; Duong, V.; Dussart, P.; Cappelle, J.; Eloit, M. A novel Polycipiviridae virus identified in Pteropus lylei stools. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef]
- Lina, P.H.C.; van Noord, M.J.; de Groot, F.G. Detection of virus in squamous papillomas of the wild bird species Fringilla coelebs. J. Natl. Cancer Inst. 1973, 50, 567–571. [Google Scholar] [CrossRef]
- Phan, T.G.; da Costa, A.C.; Zhang, W.; Pothier, P.; Ambert-Balay, K.; Deng, X.; Delwart, E. A new gyrovirus in human feces. Virus Genes 2015, 51, 132–135. [Google Scholar] [CrossRef]
- Day, J.M.; Ballard, L.L.; Duke, M.V.; Scheffler, B.E.; Zsak, L. Metagenomic analysis of the turkey gut RNA virus community. Virol. J. 2010, 7, 313. [Google Scholar] [CrossRef]
- Mihalov-Kovács, E.; Fehér, E.; Martella, V.; Bányai, K.; Farkas, S.L. The fecal virome of domesticated animals. Virus Dis. 2014, 25, 150–157. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV virus taxonomy profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Wang, H.; Yang, S.; Shan, T.; Wang, X.; Deng, X.; Delwart, E.; Zhang, W. A novel picornavirus in feces of a rainbow lorikeet (Trichoglossus moluccanus) shows a close relationship to members of the genus Avihepatovirus. Arch. Virol. 2019, 164, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- de Souza, W.M.; Fumagalli, M.J.; Martin, M.C.; de Araujo, J.; Orsi, M.A.; Sanfilippo, L.F.; Modha, S.; Durigon, E.L.; Proença-Módena, J.L.; Arns, C.W.; et al. Pingu virus: A new picornavirus in penguins from Antarctica. Virus Evol. 2019, 5, vez047. [Google Scholar] [CrossRef] [PubMed]
- Boros, Á.; Kiss, T.; Kiss, O.; Pankovics, P.; Kapusinszky, B.; Delwart, E.; Reuter, G. Genetic characterization of a novel picornavirus distantly related to the marine mammal-infecting aquamaviruses in a long-distance migrant bird species, European roller (Coracias garrulus). J. Gen. Virol. 2013, 94, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Boros, Á.; Pankovics, P.; Mátics, R.; Adonyi, Á.; Bolba, N.; Phan, T.G.; Delwart, E.; Reuter, G. Genome characterization of a novel megrivirus-related avian picornavirus from a carnivorous wild bird, western marsh harrier (Circus aeruginosus). Arch. Virol. 2017, 162, 2781–2789. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Lam, C.S.F.; Poon, R.W.S.; Tsoi, H.W.; Lee, P.; Tse, H.; Chan, A.S.L.; Luk, G.; et al. Comparative analysis of six genome sequences of three novel picornaviruses, turdiviruses 1, 2 and 3, in dead wild birds, and proposal of two novel genera, Orthoturdivirus and Paraturdivirus, in the family Picornaviridae. J. Gen. Virol. 2010, 91, 2433–2448. [Google Scholar] [CrossRef]
- Pankovics, P.; Boros, Á.; Phan, T.G.; Delwart, E.; Reuter, G. A novel passerivirus (family Picornaviridae) in an outbreak of enteritis with high mortality in estrildid finches (Uraeginthus sp.). Arch. Virol. 2018, 163, 1063–1071. [Google Scholar] [CrossRef]
- Hermanns, K.; Zirkel, F.; Kurth, A.; Drosten, C.; Junglen, S. Cimodo virus belongs to a novel lineage of reoviruses isolated from African mosquitoes. J. Gen. Virol. 2014, 95, 905–909. [Google Scholar] [CrossRef]
- Wu, N.; Zhang, P.; Liu, W.; Wang, X. Sogatella furcifera hepe-like virus: First member of a novel Hepeviridae clade identified in an insect. Virus Res. 2018, 250, 81–86. [Google Scholar] [CrossRef]
- Purdy, M.A.; Harrison, T.J.; Jameel, S.; Meng, X.J.; Okamoto, H.; van der Poel, W.H.M.; Smith, D.B. ICTV virus taxonomy profile: Hepeviridae. J. Gen. Virol. 2017, 98, 2645–2646. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Dong, X.; Hu, T.; Liu, Q.; Li, C.; Sun, Y.; Wang, Y.; Shi, W.; Zhao, Q.; Huang, J. A novel hepe-like virus from farmed giant freshwater prawn Macrobrachium rosenbergii. Viruses 2020, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral diversity of house mice in New York City. MBio 2018, 9, e01354-17. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Boros, Á.; Mátics, R.; Kapusinszky, B.; Delwart, E.; Pankovics, P. Detection and complete genome characterization of a novel RNA virus related to members of the Hepe-Virga clade in bird species, hoopoe (Upupa epops). Infect. Genet. Evol. 2020, 81, 104236. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Boros, Á.; Tóth, Z.; Kapusinszky, B.; Delwart, E.; Pankovics, P. Detection of a novel RNA virus with hepatitis E virus-like non-structural genome organization in amphibian, agile frog (Rana dalmatina) tadpoles. Infect. Genet. Evol. 2018, 65, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.R.; Hajek, A.E. Prey-processing by avian predators enhances virus transmission in the gypsy moth. Oikos 2012, 121, 1311–1316. [Google Scholar] [CrossRef]
- Koonin, E.V.; Gorbalenyat, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Family | Length (b) | Virus Name (or Closest Homolog) | Accession | Genome Region | Identity | Query Cover |
|---|---|---|---|---|---|---|---|
| dsDNA | Papillomaviridae | 407 | Francolinus leucoscepus papillomavirus 1 | YP_003104804 | L1 | 79% | 99% |
| Poxviridae | 176 | Fowlpox virus | AAZ14082 | 39 kDa core protein | 100% | 63% | |
| ssDNA | Circoviridae | 1955 | Canary circovirus | NP_573442 | Replicase | 67% | 24% |
| Parvoviridae | 346 | Goose parvovirus | ABD76400 | Nucleocapsid protein | 61% | 76% | |
| Anelloviridae | 2138 | GyV11 | MH638372 | Complete genome | - | - | |
| ssRNA (+) | Astroviridae | 6745 | PasAstV-1 | MK096773 | Complete genome | - | - |
| 6864 | PasAstV-2 | MK096774 | Complete genome | - | - | ||
| 6628 | PasAstV-3 | MK096775 | Complete genome | - | - | ||
| 6870 | PasAstV-4 | MK096776 | Complete genome | - | - | ||
| Picornaviridae | 7645 | FGPV | Complete genome | - | - |
| Group | Family | Contig Length (b) | Closest Homolog(s) | Accession | Genome Region | Identity | Query Cover |
|---|---|---|---|---|---|---|---|
| ssDNA | Poxviridae | 903 | Shearwaterpox virus | ARE67299 | Immunoglobulin domain | 45% | 27% |
| Parvoviridae | 472 | Dependoparvovirus | QHY93491 | Non-structural protein | 86% | 41% | |
| Parus major densovirus | YP_009310052 | ORF5 | 50% | 41% | |||
| ds RNA | Reoviridae | 446 | Rotavirus B | ANN82201 | RdRp | 51% | 71% |
| 283 | Rotavirus G | AXF38053 | NSP2 | 47% | 63% | ||
| 236 | Rotavirus J | APQ41756 | VP4 | 63% | 54% |
| Blastn | ||||||
|---|---|---|---|---|---|---|
| Bit Scores | Identities | |||||
| Predictors | Incidence Rate Ratios | CI | p | Estimates | CI | p |
| (Intercept) | 121.19 | 98.73–148.76 | <0.001 | 86.09 | 84.19–88.00 | <0.001 |
| site (Herreria) | 1.49 | 1.41–1.58 | <0.001 | 4.37 | 3.75–4.99 | <0.001 |
| Random Effects | ||||||
| σ2 | 0.13 | 8.60 | ||||
| τ00 | 0.30 family | 17.29 alignment_length | ||||
| 22.97 family | ||||||
| ICC | 0.69 | 0.82 | ||||
| N | 28 family | 28 family | ||||
| 367 alignment_length | ||||||
| Observations | 36,197 | 36,197 | ||||
| Marginal R2/Conditional R2 | 0.038/0.701 | 0.040/0.831 | ||||
| Blastx | ||||||
| Bit Scores | Identities | |||||
| Predictors | Estimates | CI | p | Estimates | CI | p |
| (Intercept) | 0.65 | 0.41–0.90 | <0.001 | 0.03 | −0.24–0.31 | 0.819 |
| site (Herreria) | −0.65 | −0.67–−0.62 | <0.001 | −0.27 | −0.30–−0.25 | <0.001 |
| Random Effects | ||||||
| σ2 | 0.76 | 0.48 | ||||
| τ00 | 0.58family | 0.65alignment_length | ||||
| 0.57family | ||||||
| ICC | 0.43 | 0.72 | ||||
| N | 38family | 38family | ||||
| 170alignment_length | ||||||
| Observations | 112,176 | 112,176 | ||||
| Marginal R2/Conditional R2 | 0.072/0.475 | 0.011/0.720 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Truchado, D.A.; Llanos-Garrido, A.; Oropesa-Olmedo, D.A.; Cerrada, B.; Cea, P.; Moens, M.A.J.; Gomez-Lucia, E.; Doménech, A.; Milá, B.; Pérez-Tris, J.; et al. Comparative Metagenomics of Palearctic and Neotropical Avian Cloacal Viromes Reveal Geographic Bias in Virus Discovery. Microorganisms 2020, 8, 1869. https://doi.org/10.3390/microorganisms8121869
Truchado DA, Llanos-Garrido A, Oropesa-Olmedo DA, Cerrada B, Cea P, Moens MAJ, Gomez-Lucia E, Doménech A, Milá B, Pérez-Tris J, et al. Comparative Metagenomics of Palearctic and Neotropical Avian Cloacal Viromes Reveal Geographic Bias in Virus Discovery. Microorganisms. 2020; 8(12):1869. https://doi.org/10.3390/microorganisms8121869
Chicago/Turabian StyleTruchado, Daniel A., Alejandro Llanos-Garrido, David A. Oropesa-Olmedo, Belén Cerrada, Pablo Cea, Michaël A. J. Moens, Esperanza Gomez-Lucia, Ana Doménech, Borja Milá, Javier Pérez-Tris, and et al. 2020. "Comparative Metagenomics of Palearctic and Neotropical Avian Cloacal Viromes Reveal Geographic Bias in Virus Discovery" Microorganisms 8, no. 12: 1869. https://doi.org/10.3390/microorganisms8121869
APA StyleTruchado, D. A., Llanos-Garrido, A., Oropesa-Olmedo, D. A., Cerrada, B., Cea, P., Moens, M. A. J., Gomez-Lucia, E., Doménech, A., Milá, B., Pérez-Tris, J., Cadar, D., & Benítez, L. (2020). Comparative Metagenomics of Palearctic and Neotropical Avian Cloacal Viromes Reveal Geographic Bias in Virus Discovery. Microorganisms, 8(12), 1869. https://doi.org/10.3390/microorganisms8121869