
Microbiome, Metabolome and Inflammatory Bowel Disease

Abstract
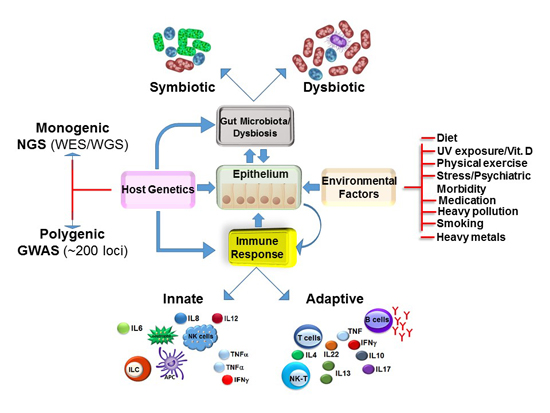
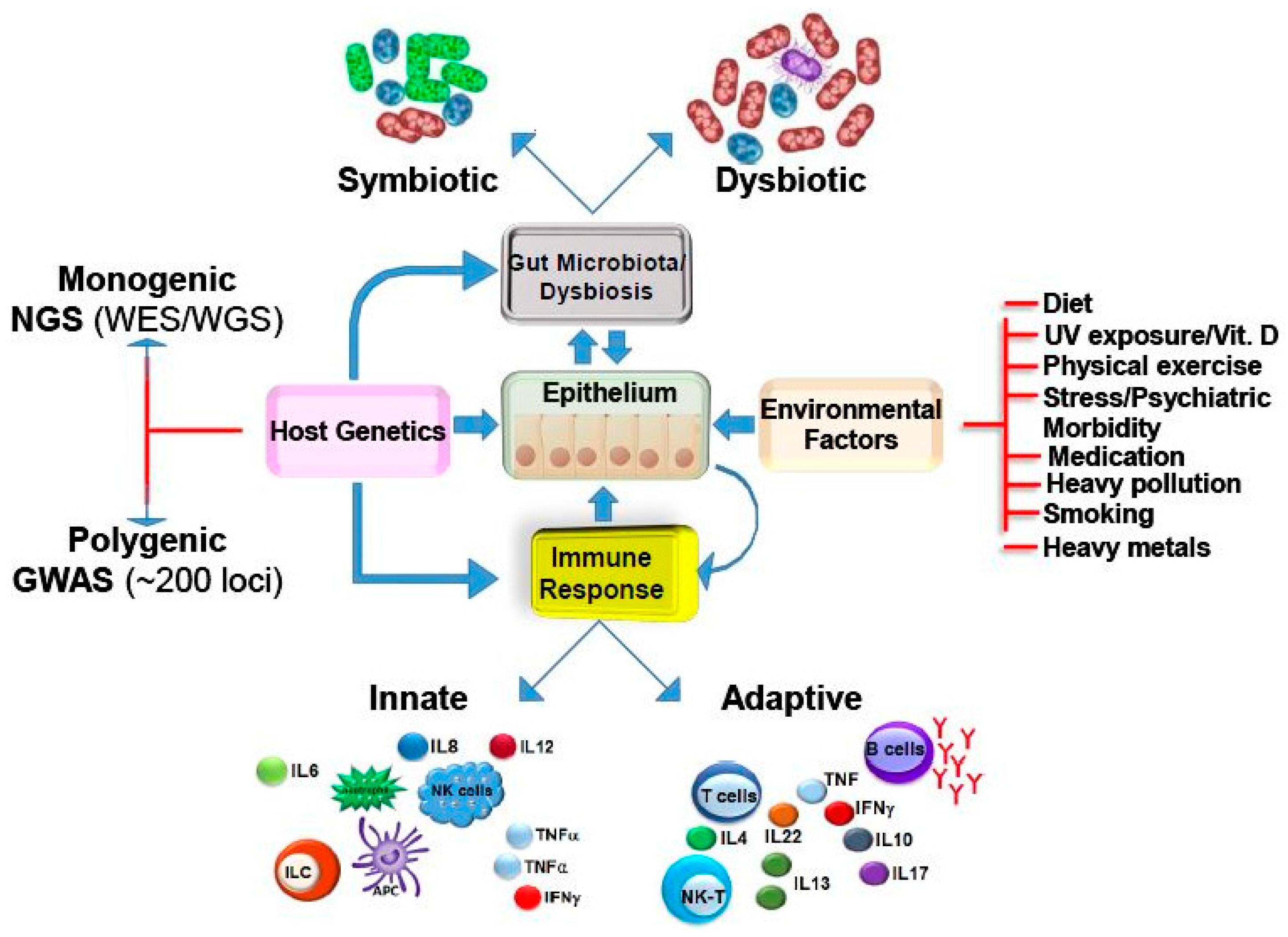
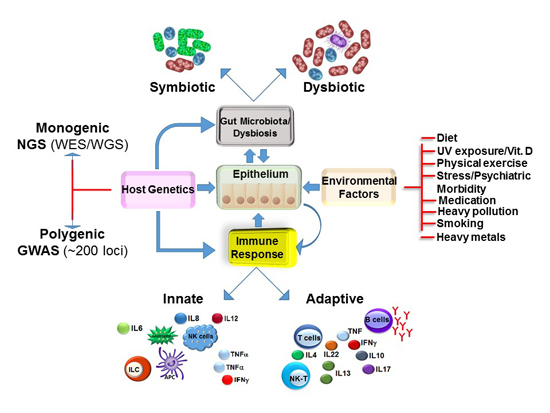
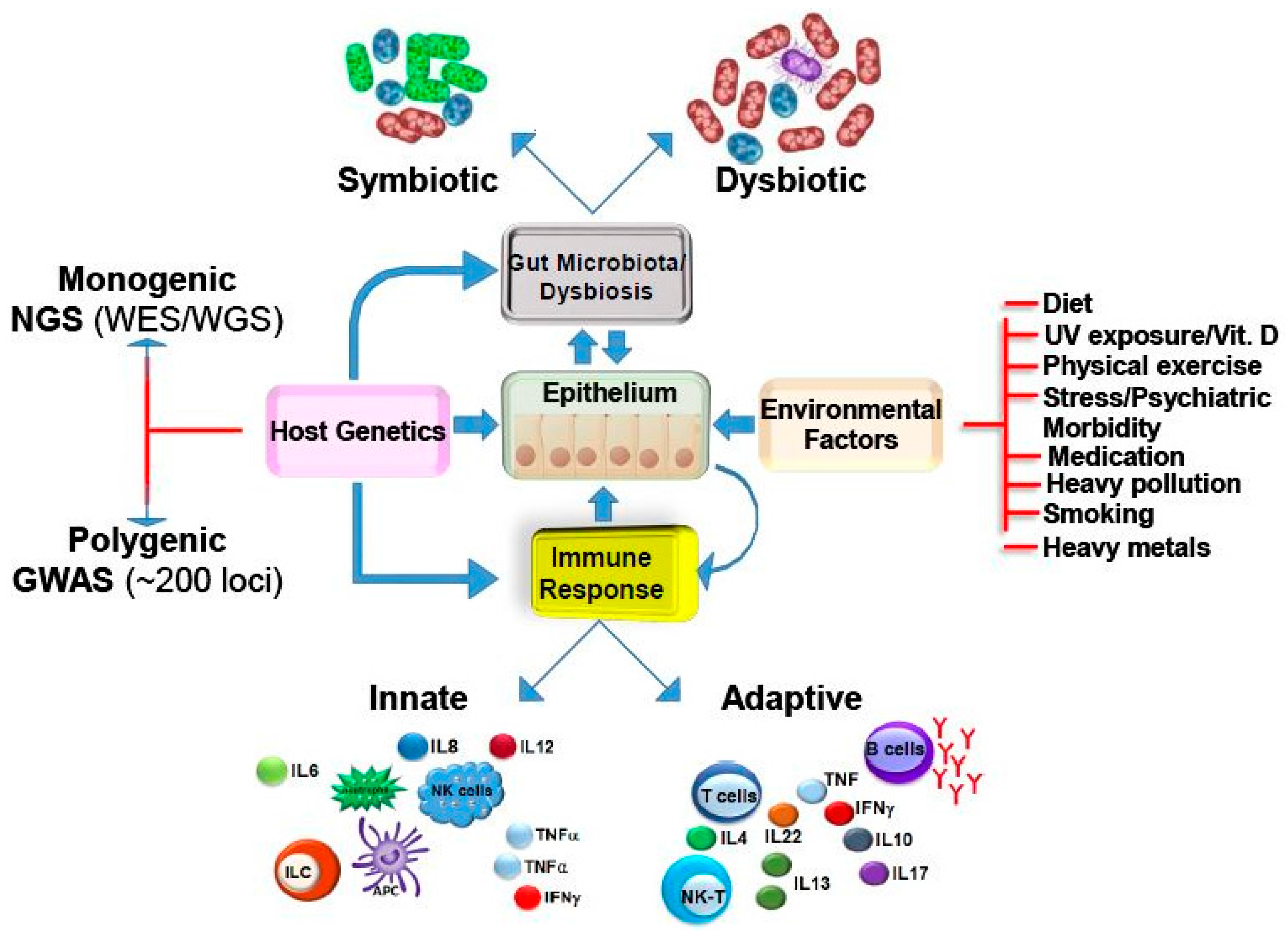
:
1. Defining Inflammatory Bowel Disease
2. Prevalence
3. Multi-Factorial Causes of IBD
4. Microbiota in Health
5. Dysbiotic and/or Pathogenic Bacteria in IBD
6. Dietary Strategies Affecting the Microbiome, Metabolome and IBD
7. Clinical Significance of Metabolomics
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eckmann, L. Animal models of inflammatory bowel disease: Lessons from enteric infections. Ann. N. Y. Acad. Sci. 2006, 1072, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Wang, L.; Shi, Y.; Cao, H.; Liu, L.; Washington, M.K.; Chaturvedi, R.; Israel, D.A.; Cao, H.; Wang, B.; et al. Berberine promotes recovery of colitis and inhibits inflammatory responses in colonic macrophages and epithelial cells in DSS-treated mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G504–G514. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef]
- Ordas, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef]
- Ferguson, L.R.; Peterman, I.; Hubner, C.; Philpott, M.; Shellin, A.N. Uncoupling gene-diet in inflammatory bowel disease (IBD). Genes Nutr. 2007, 2, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Reiff, C.; Kelly, D. Inflammatory bowel disease, gut bacteria and probiotic therapy. Int. J. Med. Microbiol. 2010, 300, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Bernstein, C.N.; Vatn, M.H.; Lakatos, P.L.; Loftus, E.V., Jr.; Tysk, C.; O’Morain, C.; Moum, B.; Colombel, J.F. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 2013, 62, 630–649. [Google Scholar] [CrossRef] [PubMed]
- Cosnes, J.; Gower-Rousseau, C.; Seksik, P.; Cortot, A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 2011, 140, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Burisch, J.; Munkholm, P. Inflammatory bowel disease epidemiology. Curr. Opin. Gastroenterol. 2013, 29, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Kappelman, M.D.; Rifas-Shiman, S.L.; Kleinman, K.; Ollendorf, D.; Bousvaros, A.; Grand, R.J.; Finkelstein, J.A. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the USA. Clin. Gastroenterol. Hepatol. 2007, 5, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Karolewska-Bochenek, K.; Lazowska-Przeorek, I.; Albrecht, P.; Grzybowska, K.; Ryzko, J.; Szamotulska, K.; Radzikowski, A.; Landowski, P.; Krzesiek, E.; Ignys, I.; et al. Epidemiology of inflammatory bowel disease among children in poland. A prospective, population-based, 2-year study, 2002–2004. Digestion 2009, 79, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.M.; Rauch, F.; Matzinger, M.A.; Benchimol, E.I.; Boland, M.; Mack, D.R. Iliac bone histomorphometry in children with newly diagnosed inflammatory bowel disease. Osteoporos. Int. 2010, 21, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Thia, K.T.; Loftus, E.V., Jr.; Sandborn, W.J.; Yang, S.K. An update on the epidemiology of inflammatory bowel disease in Asia. Am. J. Gastroenterol. 2008, 103, 3167–3182. [Google Scholar] [CrossRef] [PubMed]
- Hommes, D.; Colombel, J.F.; Emery, P.; Greco, M.; Sandborn, W.J. Changing Crohn’s disease management: Need for new goals and indices to prevent disability and improve quality of life. J. Crohns Colitis 2012, 6, S224–S234. [Google Scholar] [CrossRef]
- Kappelman, M.D.; Palmer, L.; Boyle, B.M.; Rubin, D.T. Quality of care in inflammatory bowel disease: A review and discussion. Inflamm. Bowel Dis. 2010, 16, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.B.; Ng, E.; Ozminkowski, R.J.; Wang, S.; Burton, W.N.; Goetzel, R.Z.; Maclean, R. The direct and indirect cost burden of Crohn’s disease and ulcerative colitis. J. Occup. Environ. Med. 2008, 50, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Okou, D.T.; Kugathasan, S. Role of genetics in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Fiocchi, C. Genes and ”in-vironment”: How will our concepts on the pathophysiology of inflammatory bowel disease develop in the future? Dig. Dis. 2012, 30, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Darfeuille-Michaud, A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, T.T. New cytokine targets in inflammatory bowel disease. Gastroenterol Hepatol 2011, 7, 474–476. [Google Scholar]
- Cho, J.H.; Abraham, C. Inflammatory bowel disease genetics: Nod2. Annu. Rev. Med. 2007, 58, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Orholm, M.; Binder, V.; Sorensen, T.I.; Rasmussen, L.P.; Kyvik, K.O. Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand. J. Gastroenterol. 2000, 35, 1075–1081. [Google Scholar] [PubMed]
- Halme, L.; Paavola-Sakki, P.; Turunen, U.; Lappalainen, M.; Farkkila, M.; Kontula, K. Family and twin studies in inflammatory bowel disease. World J. Gastroenterol. 2006, 12, 3668–3672. [Google Scholar] [CrossRef] [PubMed]
- Tysk, C.; Lindberg, E.; Jarnerot, G.; Floderus-Myrhed, B. Ulcerative colitis and Crohn’s disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut 1988, 29, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rotter, J.I.; Toyoda, H.; Landers, C.; Tyran, D.; McElree, C.K.; Targan, S.R. Ulcerative colitis: A genetically heterogeneous disorder defined by genetic (HLA class II) and subclinical (antineutrophil cytoplasmic antibodies) markers. J. Clin. Invest. 1993, 92, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Bethune, J.; Petersen, B.S.; Franke, A. The genetics of Crohn’s disease and ulcerative colitis —Status quo and beyond. Scand. J. Gastroenterol. 2015, 50, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.; Russell, R.K.; Satsangi, J.; Wilson, D.C. The changing epidemiology of paediatric inflammatory bowel disease. Aliment. Pharmacol. Ther. 2011, 33, 1380–1381. [Google Scholar] [CrossRef] [PubMed]
- Stappenbeck, T.S.; Rioux, J.D.; Mizoguchi, A.; Saitoh, T.; Huett, A.; Darfeuille-Michaud, A.; Wileman, T.; Mizushima, N.; Carding, S.; Akira, S.; et al. Crohn disease: A current perspective on genetics, autophagy and immunity. Autophagy 2011, 7, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Wolters, F.L.; Russel, M.G.; Sijbrandij, J.; Schouten, L.J.; Odes, S.; Riis, L.; Munkholm, P.; Langholz, E.; Bodini, P.; O’Morain, C.; et al. Disease outcome of inflammatory bowel disease patients: General outline of a Europe-wide population-based 10-year clinical follow-up study. Scand. J. Gastroenterol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Amre, D.K.; D’Souza, S.; Morgan, K.; Seidman, G.; Lambrette, P.; Grimard, G.; Israel, D.; Mack, D.; Ghadirian, P.; Deslandres, C.; et al. Imbalances in dietary consumption of fatty acids, vegetables, and fruits are associated with risk for Crohn’s disease in children. Am. J. Gastroenterol. 2007, 102, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, H.J.; Taubin, H.L. Nonsteroidal anti-inflammatory drugs activate quiescent inflammatory bowel disease. Ann. Intern. Med. 1987, 107, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Helzer, J.E. The impact of combat on later alcohol use by Vietnam veterans. J. Psychoactive Drugs 1984, 16, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Lassen, K.G.; Xavier, R.J. Advances in inflammatory bowel disease pathogenesis: Linking host genetics and the microbiome. Gut 2013, 62, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.N.; Lee, O.Y. Intestinal microbiota in pathophysiology and management of irritable bowel syndrome. World J. Gastroenterol. 2014, 20, 8886–8897. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, W.E.; Palsson, O.; Jones, K.R. Systematic review of the comorbidity of irritable bowel syndrome with other disorders: What are the causes and implications? Gastroenterology 2002, 122, 1140–1156. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. bioRxiv 2016. [Google Scholar] [CrossRef]
- Berg, R.D. The indigenous gastrointestinal microflora. Trends Microbiol. 1996, 4, 430–435. [Google Scholar] [CrossRef]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Relman, D.A. The role of microbes in Crohn’s disease. Clin. Infect. Dis. 2007, 44, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Relman, D.A. The human microbiome: Ecosystem resilience and health. Nutr. Rev. 2012, 70, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J. Microbiology: Antibiotics and adiposity. Nature 2012, 488, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Belenguer, A.; Holtrop, G.; Johnstone, A.M.; Flint, H.J.; Lobley, G.E. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol. 2007, 73, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Gratz, S.W.; Duncan, S.H.; Holtrop, G.; Ince, J.; Scobbie, L.; Duncan, G.; Johnstone, A.M.; Lobley, G.E.; Wallace, R.J.; et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 2011, 93, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Greiner, A.K.; Papineni, R.V.; Umar, S. Chemoprevention in gastrointestinal physiology and disease. Natural products and microbiome. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G1–G15. [Google Scholar] [CrossRef] [PubMed]
- Zaneveld, J.; Turnbaugh, P.J.; Lozupone, C.; Ley, R.E.; Hamady, M.; Gordon, J.I.; Knight, R. Host-bacterial coevolution and the search for new drug targets. Curr. Opin. Chem. Biol. 2008, 12, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.M.; Bast, A.; Vanhoutvin, S.A.; Fischer, M.A.; Kodde, A.; Troost, F.J.; Venema, K.; Brummer, R.J. Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. Clin. Nutr. 2009, 28, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V. Bacterial contributions to mammalian gut development. Trends Microbiol. 2004, 12, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Frick, J.S.; Autenrieth, I.B. The gut microflora and its variety of roles in health and disease. Curr. Top. Microbiol. Immunol. 2013, 358, 273–289. [Google Scholar] [PubMed]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Gosalbes, M.J.; Abellan, J.J.; Durban, A.; Perez-Cobas, A.E.; Latorre, A.; Moya, A. Metagenomics of human microbiome: Beyond 16s rDNA. Clin. Microbiol. Infect. 2012, 18, 47–49. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Li, J.V.; Marchesi, J.R.; Nicholson, J.K. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012, 16, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Comito, D.; Cascio, A.; Romano, C. Microbiota biodiversity in inflammatory bowel disease. Ital. J. Pediatr. 2014. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Targeting enteric bacteria in treatment of inflammatory bowel diseases: Why, how, and when. Curr. Opin. Gastroenterol. 2003, 19, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Taurog, J.D.; Richardson, J.A.; Croft, J.T.; Simmons, W.A.; Zhou, M.; Fernandez-Sueiro, J.L.; Balish, E.; Hammer, R.E. The germfree state prevents development of gut and joint inflammatory disease in hla-b27 transgenic rats. J. Exp. Med. 1994, 180, 2359–2364. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Sans, M.; Fiocchi, C. Inflammatory bowel disease: The role of environmental factors. Autoimmun. Rev. 2004, 3, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Kao, J.Y. The tuning of the gut nervous system by commensal microbiota. Gastroenterology 2013, 145, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Corridoni, D.; Pastorelli, L.; Mattioli, B.; Locovei, S.; Ishikawa, D.; Arseneau, K.O.; Chieppa, M.; Cominelli, F.; Pizarro, T.T. Probiotic bacteria regulate intestinal epithelial permeability in experimental ileitis by a TNF-dependent mechanism. PLoS ONE 2012, 7, e42067. [Google Scholar] [CrossRef] [PubMed]
- Gionchetti, P.; Rizzello, F.; Habal, F.; Morselli, C.; Amadini, C.; Romagnoli, R.; Campieri, M. Standard treatment of ulcerative colitis. Dig. Dis. 2003, 21, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Thomson, J.M.; El-Omar, E.M.; Hold, G.L. The role of infection in the aetiology of inflammatory bowel disease. J. Gastroenterol. 2010, 45, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Lepage, P.; Seksik, P.; Dore, J.; Marteau, P. Temperature gradient gel electrophoresis of fecal 16s rRNA reveals active Escherichia coli in the microbiota of patients with ulcerative colitis. J. Clin. Microbiol. 2006, 44, 3172–3177. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Mondot, S.; Kang, S.; Furet, J.P.; Aguirre de Carcer, D.; McSweeney, C.; Morrison, M.; Marteau, P.; Dore, J.; Leclerc, M. Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm. Bowel Dis. 2011, 17, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Huys, G.; Cnockaert, M.; de Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Prindiville, T.P.; Sheikh, R.A.; Cohen, S.H.; Tang, Y.J.; Cantrell, M.C.; Silva, J., Jr. Bacteroides fragilis enterotoxin gene sequences in patients with inflammatory bowel disease. Emerg. Infect. Dis. 2000, 6, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Neut, C.; Bulois, P.; Desreumaux, P.; Membre, J.M.; Lederman, E.; Gambiez, L.; Cortot, A.; Quandalle, P.; van Kruiningen, H.; Colombel, J.F. Changes in the bacterial flora of the neoterminal ileum after ileocolonic resection for Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Kuzuoka, H.; Tsujikawa, T.; Nakamura, S.; Hirai, F.; Suzuki, Y.; Matsui, T.; Fujiyama, Y.; Matsumoto, T. Multicenter analysis of fecal microbiota profiles in Japanese patients with Crohn’s disease. J. Gastroenterol. 2012, 47, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Seksik, P.; Rigottier-Gois, L.; Gramet, G.; Sutren, M.; Pochart, P.; Marteau, P.; Jian, R.; Dore, J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 2003, 52, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Lucke, K.; Miehlke, S.; Jacobs, E.; Schuppler, M. Prevalence of bacteroides and Prevotella spp. In ulcerative colitis. J. Med. Microbiol. 2006, 55, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhya, I.; Hansen, R.; El-Omar, E.M.; Hold, G.L. IBD-what role do proteobacteria play? Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Lepage, P.; Nolte, A.; Hellmig, S.; Schreiber, S.; Ott, S.J. Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J. Med. Microbiol. 2010, 59, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, M.; Dogan, B.; Rishniw, M.; Weitzman, G.; Bosworth, B.; Yantiss, R.; Orsi, R.H.; Wiedmann, M.; McDonough, P.; Kim, S.G.; et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive escherichia coli of novel phylogeny relative to depletion of clostridiales in crohn’s disease involving the ileum. ISME J. 2007, 1, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Gophna, U.; Sommerfeld, K.; Gophna, S.; Doolittle, W.F.; Veldhuyzen van Zanten, S.J. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J. Clin. Microbiol. 2006, 44, 4136–4141. [Google Scholar] [CrossRef] [PubMed]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of enterobacteriaceae. Cell Host Microbe 2007, 2, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Willing, B.; Halfvarson, J.; Dicksved, J.; Rosenquist, M.; Jarnerot, G.; Engstrand, L.; Tysk, C.; Jansson, J.K. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Boudeau, J.; Glasser, A.L.; Masseret, E.; Joly, B.; Darfeuille-Michaud, A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect. Immun. 1999, 67, 4499–4509. [Google Scholar] [PubMed]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Naser, S.A.; Sagramsingh, S.R.; Naser, A.S.; Thanigachalam, S. Mycobacterium avium subspecies paratuberculosis causes Crohn’s disease in some inflammatory bowel disease patients. World J. Gastroenterol. 2014, 20, 7403–7415. [Google Scholar] [CrossRef] [PubMed]
- Nazareth, N.; Magro, F.; Machado, E.; Ribeiro, T.G.; Martinho, A.; Rodrigues, P.; Alves, R.; Macedo, G.N.; Gracio, D.; Coelho, R.; et al. Prevalence of Mycobacterium avium subsp. paratuberculosis and Escherichia coli in blood samples from patients with inflammatory bowel disease. Med. Microbiol. Immunol. 2015, 204, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Mahendran, V.; Riordan, S.M.; Grimm, M.C.; Tran, T.A.; Major, J.; Kaakoush, N.O.; Mitchell, H.; Zhang, L. Prevalence of campylobacter species in adult Crohn’s disease and the preferential colonization sites of campylobacter species in the human intestine. PLoS ONE 2011, 6, e25417. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Zhang, L.; Day, A.S.; Leach, S.T.; Lemberg, D.A.; Mitchell, H. Campylobacter concisus and other campylobacter species in children with newly diagnosed Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Man, S.M.; Day, A.S.; Leach, S.T.; Lemberg, D.A.; Dutt, S.; Stormon, M.; Otley, A.; O’Loughlin, E.V.; Magoffin, A.; et al. Detection and isolation of campylobacter species other than C. jejuni from children with Crohn’s disease. J. Clin. Microbiol. 2009, 47, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Rhee, S.H.; Pothoulakis, C.; Lamont, J.T. Inflammation and apoptosis in clostridium difficile enteritis is mediated by PGE2 up-regulation of Fas ligand. Gastroenterology 2007, 133, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.; Vijayapal, A.; Graham, M.B.; Beaulieu, D.B.; Otterson, M.F.; Lundeen, S.; Skaros, S.; Weber, L.R.; Komorowski, R.A.; Knox, J.F.; et al. Impact of clostridium difficile on inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2007, 5, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Rabizadeh, S.; Rhee, K.J.; Wu, S.; Huso, D.; Gan, C.M.; Golub, J.E.; Wu, X.; Zhang, M.; Sears, C.L. Enterotoxigenic Bacteroides fragilis: A potential instigator of colitis. Inflamm. Bowel Dis. 2007, 13, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Blaser, M.J.; Ley, R.E.; Knight, R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology 2011, 140, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [PubMed]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 4578–4585. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Friedberg, I.; Ivanov, I.V.; Davidson, L.A.; Goldsby, J.S.; Dahl, D.B.; Herman, D.; Wang, M.; Donovan, S.M.; Chapkin, R.S. A metagenomic study of diet-dependent interaction between gut microbiota and host in infants reveals differences in immune response. Genome Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.C.; Costabile, A.; Holland, D.T.; Hoyles, L.; McCartney, A.L. Examination of faecal bifidobacterium populations in breast- and formula-fed infants during the first 18 months of life. Microbiology 2010, 156, 3329–3341. [Google Scholar] [CrossRef] [PubMed]
- Fallani, M.; Amarri, S.; Uusijarvi, A.; Adam, R.; Khanna, S.; Aguilera, M.; Gil, A.; Vieites, J.M.; Norin, E.; Young, D.; et al. Determinants of the human infant intestinal microbiota after the introduction of first complementary foods in infant samples from five European centres. Microbiology 2011, 157, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Fallani, M.; Young, D.; Scott, J.; Norin, E.; Amarri, S.; Adam, R.; Aguilera, M.; Khanna, S.; Gil, A.; Edwards, C.A.; et al. Intestinal microbiota of 6-week-old infants across Europe: Geographic influence beyond delivery mode, breast-feeding, and antibiotics. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Chapman-Kiddell, C.A.; Davies, P.S.; Gillen, L.; Radford-Smith, G.L. Role of diet in the development of inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.K.; Abraham, B.; El-Serag, H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am. J. Gastroenterol. 2011, 106, 563–573. [Google Scholar] [CrossRef] [PubMed]
- D’Haens, G.R.; Sartor, R.B.; Silverberg, M.S.; Petersson, J.; Rutgeerts, P. Future directions in inflammatory bowel disease management. J. Crohns Colitis 2014, 8, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Richman, E.; Rhodes, J.M. Review article: Evidence-based dietary advice for patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2013, 38, 1156–1171. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Spooren, C.E.; Pierik, M.J.; Zeegers, M.P.; Feskens, E.J.; Masclee, A.A.; Jonkers, D.M. Review article: The association of diet with onset and relapse in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2013, 38, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.K.; Lee, D.; Lewis, J. Diet and inflammatory bowel disease: Review of patient-targeted recommendations. Clin. Gastroenterol. Hepatol. 2014, 12, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Environmental risk factors for inflammatory bowel disease. Gastroenterol. Hepatol. 2013, 9, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Durchschein, F.; Petritsch, W.; Hammer, H.F. Diet therapy for inflammatory bowel diseases: The established and the new. World J. Gastroenterol. 2016, 22, 2179–2194. [Google Scholar] [PubMed]
- Walton, C.; Montoya, M.P.; Fowler, D.P.; Turner, C.; Jia, W.; Whitehead, R.N.; Griffiths, L.; Waring, R.H.; Ramsden, D.B.; Cole, J.A.; et al. Enteral feeding reduces metabolic activity of the intestinal microbiome in Crohn’s disease: An observational study. Eur. J. Clin. Nutr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kaakoush, N.O.; Day, A.S.; Leach, S.T.; Lemberg, D.A.; Nielsen, S.; Mitchell, H.M. Effect of exclusive enteral nutrition on the microbiota of children with newly diagnosed Crohn’s disease. Clin. Transl. Gastroenterol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Day, A.S.; Lopez, R.N. Exclusive enteral nutrition in children with Crohn’s disease. World J. Gastroenterol. 2015, 21, 6809–6816. [Google Scholar] [PubMed]
- Heuschkel, R.B.; Menache, C.C.; Megerian, J.T.; Baird, A.E. Enteral nutrition and corticosteroids in the treatment of acute Crohn’s disease in children. J. Pediatr. Gastroenterol. Nutr. 2000, 31, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Afzal, N.A.; Davies, S.; Paintin, M.; Arnaud-Battandier, F.; Walker-Smith, J.A.; Murch, S.; Heuschkel, R.; Fell, J. Colonic Crohn’s disease in children does not respond well to treatment with enteral nutrition if the ileum is not involved. Dig. Dis. Sci. 2005, 50, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Li, P.; Xu, L.; Peng, Z.; Xiang, J.; He, Z.; Zhang, T.; Ji, G.; Nie, Y.; Wu, K.; et al. Step-up fecal microbiota transplantation (FMT) strategy. Gut Microbes 2016. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.K.; El Kasmi, K.C.; Anderson, A.L.; Devereaux, M.W.; Fillon, S.A.; Robertson, C.E.; Wagner, B.D.; Stevens, M.J.; Pace, N.R.; Sokol, R.J. Specific microbiome changes in a mouse model of parenteral nutrition associated liver injury and intestinal inflammation. PLoS ONE 2014, 9, e110396. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Chang, E.B. Diet-induced expansion of pathobionts in experimental colitis: Implications for tailored therapies. Gut Microbes 2013, 4, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct Genomics 2001, 2, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.L.; Nicholls, A.W. Metabolomics as a functional genomic tool for understanding lipid dysfunction in diabetes, obesity and related disorders. Pharmacogenomics 2006, 7, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ”Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Maurice, C.F.; Haiser, H.J.; Turnbaugh, P.J. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 2013, 152, 39–50. [Google Scholar] [CrossRef] [PubMed]
- McNulty, N.P.; Yatsunenko, T.; Hsiao, A.; Faith, J.J.; Muegge, B.D.; Goodman, A.L.; Henrissat, B.; Oozeer, R.; Cools-Portier, S.; Gobert, G.; et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Marcobal, A.; Kashyap, P.C.; Nelson, T.A.; Aronov, P.A.; Donia, M.S.; Spormann, A.; Fischbach, M.A.; Sonnenburg, J.L. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. Isme J. 2013, 7, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Bamba, T.; Harada, K.; Fukusaki, E.; Kobayashi, A. Time-course metabolic profiling in Arabidopsis thaliana cell cultures after salt stress treatment. J. Exp. Bot. 2007, 58, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Borner, J.; Buchinger, S.; Schomburg, D. A high-throughput method for microbial metabolome analysis using gas chromatography/mass spectrometry. Anal. Biochem. 2007, 367, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Tolstikov, V.; Fiehn, O.; Weiss, R.H. A comprehensive urinary metabolomic approach for identifying kidney cancerr. Anal. Biochem. 2007, 363, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Parveen, I.; Moorby, J.M.; Fraser, M.D.; Allison, G.G.; Kopka, J. Application of gas chromatography-mass spectrometry metabolite profiling techniques to the analysis of heathland plant diets of sheep. J. Agric. Food Chem. 2007, 55, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Haiser, H.J.; Turnbaugh, P.J. Is it time for a metagenomic basis of therapeutics? Science 2012, 336, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, Y.; Nishiumi, S.; Ooi, M.; Hatano, N.; Shinohara, M.; Yoshie, T.; Kondo, Y.; Furumatsu, K.; Shiomi, H.; Kutsumi, H.; et al. Gcms-based metabolomic study in mice with colitis induced by dextran sulfate sodium. Inflamm. Bowel Dis. 2011, 17, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Dawiskiba, T.; Deja, S.; Mulak, A.; Zabek, A.; Jawien, E.; Pawelka, D.; Banasik, M.; Mastalerz-Migas, A.; Balcerzak, W.; Kaliszewski, K.; et al. Serum and urine metabolomic fingerprinting in diagnostics of inflammatory bowel diseases. World J. Gastroenterol. 2014, 20, 163–174. [Google Scholar] [CrossRef] [PubMed]
- De Preter, V.; Machiels, K.; Joossens, M.; Arijs, I.; Matthys, C.; Vermeire, S.; Rutgeerts, P.; Verbeke, K. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 2015, 64, 447–458. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, K.; Reed, D.E.; Schneider, T.; Dang, F.; Keshteli, A.H.; de Palma, G.; Madsen, K.; Bercik, P.; Vanner, S. Fodmaps alter symptoms and the metabolome of patients with IBS: A randomised controlled trial. Gut 2016. [Google Scholar] [CrossRef] [PubMed]
- Machiels, K.; Joossens, M.; Sabino, J.; de Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased proportions of bifidobacterium and the lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Drugging the gut microbiome. Nat. Biotechnol. 2015, 33, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Spiller, R. Irritable bowel syndrome: New insights into symptom mechanisms and advances in treatment. F1000Research 2016. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, K.; Kumar, S.; Singh, R.R.; Sharma, U.; Ahuja, V.; Makharia, G.K.; Jagannathan, N.R. Metabolism of the colonic mucosa in patients with inflammatory bowel diseases: An in vitro proton magnetic resonance spectroscopy study. Magn. Reson. Imaging 2009, 27, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Bezabeh, T.; Somorjai, R.L.; Smith, I.C.; Nikulin, A.E.; Dolenko, B.; Bernstein, C.N. The use of 1H magnetic resonance spectroscopy in inflammatory bowel diseases: Distinguishing ulcerative colitis from Crohn’s disease. Am. J. Gastroenterol. 2001, 96, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.P.; Mortensen, N.J. Ileal pouch surgery for ulcerative colitis. World J. Gastroenterol. 2007, 13, 3288–3300. [Google Scholar] [CrossRef] [PubMed]
- Odze, R.D. A contemporary and critical appraisal of ”indeterminate colitis”. Mod. Pathol. 2015, 28, S30–S46. [Google Scholar] [CrossRef] [PubMed]
- Tontini, G.E.; Vecchi, M.; Pastorelli, L.; Neurath, M.F.; Neumann, H. Differential diagnosis in inflammatory bowel disease colitis: State of the art and future perspectives. World J. Gastroenterol. 2015, 21, 21–46. [Google Scholar] [CrossRef] [PubMed]
- Dotan, I. New serologic markers for inflammatory bowel disease diagnosis. Dig. Dis. 2010, 28, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Van Schaik, F.D.; Oldenburg, B.; Hart, A.R.; Siersema, P.D.; Lindgren, S.; Grip, O.; Teucher, B.; Kaaks, R.; Bergmann, M.M.; Boeing, H.; et al. Serological markers predict inflammatory bowel disease years before the diagnosis. Gut 2013, 62, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Zholudev, A.; Zurakowski, D.; Young, W.; Leichtner, A.; Bousvaros, A. Serologic testing with ANCA, ASCA, and anti-OmpC in children and young adults with Crohn’s disease and ulcerative colitis: Diagnostic value and correlation with disease phenotype. Am. J. Gastroenterol. 2004, 99, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Kuna, A.T. Serological markers of inflammatory bowel disease. Biochem. Med. 2013, 23, 28–42. [Google Scholar] [CrossRef]
- Schicho, R.; Shaykhutdinov, R.; Ngo, J.; Nazyrova, A.; Schneider, C.; Panaccione, R.; Kaplan, G.G.; Vogel, H.J.; Storr, M. Quantitative metabolomic profiling of serum, plasma, and urine by 1H NMR spectroscopy discriminates between patients with inflammatory bowel disease and healthy individuals. J. Proteome Res. 2012, 11, 3344–3357. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.R.; Willsmore, J.D.; Cox, I.J.; Walker, D.G.; Cobbold, J.F.; Taylor-Robinson, S.D.; Orchard, T.R. Serum metabolic profiling in inflammatory bowel disease. Dig. Dis. Sci. 2012, 57, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.S.; Siffledeen, J.; Su, X.; Murdoch, T.B.; Fedorak, R.N.; Slupsky, C.M. Urinary NMR metabolomic profiles discriminate inflammatory bowel disease from healthy. J. Crohns Colitis 2013, 7, e42–e48. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, G.; Noor, S.O.; Ridgway, K.; Scovell, L.; Jamieson, C.; Johnson, I.T.; Colquhoun, I.J.; Kemsley, E.K.; Narbad, A. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J. Proteome Res. 2011, 10, 4208–4218. [Google Scholar] [CrossRef] [PubMed]
- Bjerrum, J.T.; Nielsen, O.H.; Hao, F.; Tang, H.; Nicholson, J.K.; Wang, Y.; Olsen, J. Metabonomics in ulcerative colitis: Diagnostics, biomarker identification, and insight into the pathophysiology. J. Proteome Res. 2010, 9, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.R.; Cox, I.J.; Walker, D.G.; North, B.V.; Patel, V.M.; Marshall, S.E.; Jewell, D.P.; Ghosh, S.; Thomas, H.J.; Teare, J.P.; et al. Characterization of inflammatory bowel disease with urinary metabolic profiling. Am. J. Gastroenterol. 2009, 104, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Jansson, J.; Willing, B.; Lucio, M.; Fekete, A.; Dicksved, J.; Halfvarson, J.; Tysk, C.; Schmitt-Kopplin, P. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS ONE 2009, 4, e6386. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Rigottier-Gois, L.; Lay, C.; Lepage, P.; Podglajen, I.; Marteau, P.; Dore, J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Therapeutic correction of bacterial dysbiosis discovered by molecular techniques. Proc. Natl. Acad. Sci. USA 2008, 105, 16413–16414. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.R.; Holmes, E.; Khan, F.; Kochhar, S.; Scanlan, P.; Shanahan, F.; Wilson, I.D.; Wang, Y. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J. Proteome Res. 2007, 6, 546–551. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| S. No | Increased | Decreased | Increased | Decreased |
|---|---|---|---|---|
| 1 | Phylum proteobacteria [78,81] | Phylum Firmicutes [70,71,72,160] | Colon mucosal tissue CD: glucose, glycerophosphorylcholine UC: arginine, glucose, glycerophosphorylcholine, lysine [144] | Colon mucosal tissue CD: alanine, choline, formate, glutamine/glutamate, isoleucine/leucine/valine, lactate, myoinositol, succinate UC: alanine, choline, formate, glutamine/glutamate, isoleucine/leucine/valine, lactate, myoinositol, succinate [144] |
| 2 | Adherent-invasive E. coli (AIEC) [87], Campylobacter concisus [80], Clostridium difficil) [92], Bacteroides fragilis [75], Bacteroides vulgatus, Klebssiella pneumonie, fusobacterium varium [161]) | Butyrate producing bacteria e.g., Roseburia hominis and Faecalibacterium [136] | Fecal matter CD: alanine, glycerol, isoleucine, leucine, lysine, valine UC: glutamate, lysine [162] | Fecal matter CD: acetate, butyrate, methylamine, Trimethylamine UC: methylamine, trimethylamine [162] |
| 3 | R. gnavus [74] | Microbial diversity [69] | Urine CD: formate, glycine, glycolate, guanidoacetate, methylhistidine UC: citrate, glycine, glycolate, guanidoacetate, methylhistidine [158] | Urine CD: 4-cresol sulfate, citrate, hippurate UC: hippurate, trimethyllysine [158] |
| 4 | CD:Mycobacterium avium paratuberculosis (MAP) [72] | Microbial genes in feces [43] | ND | SCFA synthesis [136] |
| 5 | Enterotoxigenic B. fragilis (ETBF) [98] | Decreased presence of anti-inflammatory F. prausnitzii, B. adolescentis, D. invisus [74] | ND | Amino acid biosynthesis [147] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. https://doi.org/10.3390/microorganisms4020020
Ahmed I, Roy BC, Khan SA, Septer S, Umar S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms. 2016; 4(2):20. https://doi.org/10.3390/microorganisms4020020
Chicago/Turabian StyleAhmed, Ishfaq, Badal C. Roy, Salman A. Khan, Seth Septer, and Shahid Umar. 2016. "Microbiome, Metabolome and Inflammatory Bowel Disease" Microorganisms 4, no. 2: 20. https://doi.org/10.3390/microorganisms4020020
APA StyleAhmed, I., Roy, B. C., Khan, S. A., Septer, S., & Umar, S. (2016). Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms, 4(2), 20. https://doi.org/10.3390/microorganisms4020020