
Metagenomic Analyses Reveal That Energy Transfer Gene Abundances Can Predict the Syntrophic Potential of Environmental Microbial Communities



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Selection of Metagenomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Metagenome Name | IMG Genome ID | Source | Gene Count | Classification |
|---|---|---|---|---|---|
| 1 | Marine microbial communities from Deepwater Horizon subsurface plume in Gulf of Mexico, 16-4 Below Plume (16-4 Below Plume) | 3300005379 | IMG | 112790 | Non-Syntrophic Hydrocarbon |
| 2 | Marine microbial communities from Deepwater Horizon subsurface plume in Gulf of Mexico, 16-5 In Plume (16-5 In Plume) | 3300005380 | IMG | 114085 | Non-Syntrophic Hydrocarbon |
| 3 | Marine microbial communities from Deepwater Horizon subsurface plume in Gulf of Mexico, 52-1 Below Plume (52-1 Below Plume) | 2149837027 | IMG | 60113 | Non-Syntrophic Hydrocarbon |
| 4 | Marine microbial communities from Deepwater Horizon subsurface plume in Gulf of Mexico, 52-4 In plume (52-4 In Plume) | 3300005378 | IMG | 102800 | Non-Syntrophic Hydrocarbon |
| 5 | Oil sands microbial communities from Horse River, Alberta, Canada—outcrops (H1C: 454 sequencing assembly) | 3300001422 | HMP | 570427 | Non-Syntrophic Hydrocarbon |
| 6 | Oil sands microbial communities from Horse River, Alberta, Canada—outcrops collected from inside the river (H1R: 454 sequencing assembly) | 3300001393 | HMP | 559882 | Non-Syntrophic Hydrocarbon |
| 7 † | Syncrude MLSB Tailings Pond Water Surface—Tailings pond microbial communities from Northern Alberta—Syncrude Mildred Lake Settling Basin (PDSYNTPWS: 454+illumina sequencing assembly) | 3300001605 | HMP | 3740874 | Non-Syntrophic Hydrocarbon |
| 8 | Syncrude MLSB WIP Surface + Isolates—Tailings pond microbial communities from Northern Alberta—Syncrude Mildred Lake Settling Basin (WIP-PD_SYN_TP_WS_002_003_071511 and isolates PD8, PD9 joint assembly) | 3300001239 | HMP | 768174 | Non-Syntrophic Hydrocarbon |
| 9 | Wastewater microbial communities from Syncrude, Ft. McMurray, Alberta—Microbes from Oil-contaminated ecosystem in Alberta, Canada Inniskillen 604.3 (Inniskillen 604.3: 454 sequencing assembly) | 3300001190 | HMP | 25694 | Non-Syntrophic Hydrocarbon |
| 10 | Wastewater microbial communities from Syncrude, Ft. McMurray, Alberta—Microbes from Sediment core from a heavy oil reservoir, Alberta Canada Inniskillen 614.3 (Inniskillen 614.3: 454+illumina sequencing assembly) | 3300001197 | HMP | 204944 | Non-Syntrophic Hydrocarbon |
| 11 | Arctic peat soil from Barrow, Alaska—NGEE Surface sample 210-1 shallow-072012 (NGEE Surface sample 210-1 shallow-072012, ASSEMBLY_DATE=20130514) | 3300001414 | IMG | 14379538 | Non-Syntrophic |
| 12 | Arctic peat soil from Barrow, Alaska—NGEE Surface sample 210-2 deep-072012 (NGEE Surface sample 210-2 deep-072012, ASSEMBLY_DATE=20130514) | 3300001396 | IMG | 8697097 | Non-Syntrophic |
| 13 | Arctic peat soil from Barrow, Alaska—NGEE Surface sample 210-2 deep-092012 (NGEE Surface sample 210-2 deep-092012, ASSEMBLY_DATE=20130516) | 3300001385 | IMG | 6241552 | Non-Syntrophic |
| 14 | Arctic peat soil from Barrow, Alaska—NGEE Surface sample 210-2 shallow-072012 (NGEE Surface sample 210-2 shallow-072012, ASSEMBLY_DATE=20130514) | 3300001416 | IMG | 14687361 | Non-Syntrophic |
| 15 | Freshwater microbial communities from Lake Mendota, WI—02JUN2012 deep hole epilimnion (Lake Mendota Deep Hole Epilimnion 02Jun12, ASSEMBLY_DATE=20140125) | 3300002296 | IMG | 1990049 | Non-Syntrophic |
| 16 | Human retroauricular crease microbial communities from NIH, USA—visit 1, subject 338793263 | 7000000458 | IMG | 36795 | Non-Syntrophic |
| 17 | Human right retroauricular crease microbial communities from NIH, USA—visit 2 of subject 763961826 replicate 2 | 7000000031 | IMG | 39970 | Non-Syntrophic |
| 18 | Marine microbial communities from expanding oxygen minimum zones in Line P, North Pacific Ocean—August 2009 P16 10m (Line P August 2009 P16 10m, March 2012 Assem) | 3300000149 | IMG | 238270 | Non-Syntrophic |
| 19 | Marine microbial communities from expanding oxygen minimum zones in Line P, North Pacific Ocean—June 2008 P4 1300m (Line P June 2008 P4 1300m, March 2012 Assem) | 3300000141 | IMG | 256292 | Non-Syntrophic |
| 20 | Marine microbial communities from expanding oxygen minimum zones in the Saanich Inlet—54 02/08/11 120m (Saanich Inlet 54 02/08/11 120m, March 2012 Assem) | 3300000146 | IMG | 169407 | Non-Syntrophic |
| 21 | Soil microbial communities from Great Prairies—Iowa, Native Prairie soil (Iowa, Native Prairie soil, Feb 2012 Assem MSU hiseq + gaii) | 3300000364 | IMG | 8508638 | Non-Syntrophic |
| 22 | Subsurface groundwater microbial communities from S. Glens Falls, New York, USA—GMW60B uncontaminated upgradient, 5.4 m (Subsurface groundwater monitoring well GMW60B uncontam upgr,5.4m, Oct 2012 Assem) | 3300000571 | IMG | 1391570 | Non-Syntrophic |
| 23 | Switchgrass and industrial compost incubating bioreactor microbial communities from the Joint BioEnergy Institute, California, USA, that is aerobic and thermophilic—SG0.5JP960 (454-Illumina assembly) —version 2 (454-Illumina assembly v2) | 3300005442 | IMG | 62968 | Non-Syntrophic |
| 24 | Wastewater treatment Type I Accumulibacter community from EBPR Bioreactor in Madison, WI—N_134min_Aerobic | 3300002344 | IMG | 367402 | Non-Syntrophic |
| 25 | Coal bed methane well microbial communities from Alberta, Canada (CO182: coal bed cutting Illumina Assembly) | 3300000052 | HMP | 665055 | Other |
| 26 | Coal-degrading lab enrichment microbial communities from Bowden, Alberta, Canada—QSAFCN5 (QSAFCN5: 454 assembly) | 3300000507 | HMP | 257387 | Other |
| 27 | Coal-degrading lab enrichment microbial communities from Bowden, Alberta, Canada—methanogenic culture: QSAFCN2 (QSAFCN2 454 assembly) | 3300000408 | HMP | 223117 | Other |
| 43 | Sheep rumen microbial communities from New Zealand—Rank43_high (high_rank43) | 3300001531 | IMG | 911348 | Other |
| 28 | Benzene-degrading bioreactor microbial communities from Toronto, Ontario, Canada, that are methanogenic—September 2009 gDNA_4 (Assembly with PE data) | 2061766000 | IMG | 207753 | Syntrophic Culture |
| 29 | Hydrocarbon resource environments microbial communities from Canada and USA—Toluene degrading community from Alberta, Canada (Toluene: 454+illumina+illuminaFosmid sequencing assembly) | 3300001567 | HMP | 1184637 | Syntrophic Culture |
| 30 | Oil sands microbial community from Northern Alberta which degrade Naphthaline (NapDC: 454 and illumina hybrid assembly) | 3300000032 | HMP | 749231 | Syntrophic Culture |
| 31 | Tailings pond microbial communities from Northern Alberta—Short chain hydrocarbon degrading methanogenic enrichment culture SCADC: (SCADC: 454+illumina assembly) | 3300000568 | HMP | 1513645 | Syntrophic Culture |
| 32 | Wastewater bioreactor microbial communities from Singapore—TA reactor DNA contigs from 4 sample (re-annotation) (MER-FS) (assembled) | 3300005443 | IMG | 95700 | Syntrophic Culture |
| 33 | Subsurface groundwater microbial communities from S. Glens Falls, New York, USA—GMW46 contaminated, 5.4 m (Subsurface groundwater monitoring well GMW46 contaminated, 5.4m, Oct 2012 Assem) | 3300000574 | IMG | 1250031 | Potentially Syntrophic Hydrocarbon |
| 34 | Tailings pond microbial communities from Northern Alberta—TP6_2008_30ft: | 2228664008 | HMP | 389458 | Potentially Syntrophic Hydrocarbon |
| 35 | Tar lake microbial communities from La Brea, Trinidad and Tobago | 2228664012 | HMP | 195772 | Potentially Syntrophic Hydrocarbon |
| 36 | Wastewater microbial communities from Syncrude, Ft. McMurray, Alberta—Microbes from Suncor taillings pond 6 2012TP6_6 (2012TP6_6m: illumina sequencing assembly) | 3300001580 | HMP | 3205687 | Potentially Syntrophic Hydrocarbon |
| 37 | Wastewater microbial communities from Syncrude, Ft. McMurray, Alberta—Microbes in water sample from Medicine Hat oil field -PW_MHGC_2012April2: (PW_MHGC_2012April2: 454+illumina sequencing assembly final) | 3300001444 | HMP | 2443755 | Potentially Syntrophic Hydrocarbon |
| 38 | Wastewater microbial communities from Syncrude, Ft. McMurray, Alberta—Microbes in water sample from Medicine Hat oil field -PW_MHGC_2012April2: (PW_MHGC_2012April2: 454 + illumina sequencing assembly) | 3300001592 | HMP | 2251644 | Potentially Syntrophic Hydrocarbon |
| 39 | Tailings Pipe from MSLB 2011 -Wastewater microbial communities from Syncrude, Ft. McMurray, Alberta—West In Pit SyncrudeMLSB2011 (SyncrudeMLSB2011: 454+illumina assembly) | 3300000558 | HMP | 3021632 | Potentially Syntrophic Hydrocarbon |
| 42 | Marine sediment microbial communities from Arctic Ocean, off the coast from Alaska—sample from high methane PC12-225-485cm (High methane PC12-225-485cm Jan 2011 assembly) | 2140918005 | IMG | 674403 | Potentially Syntrophic |
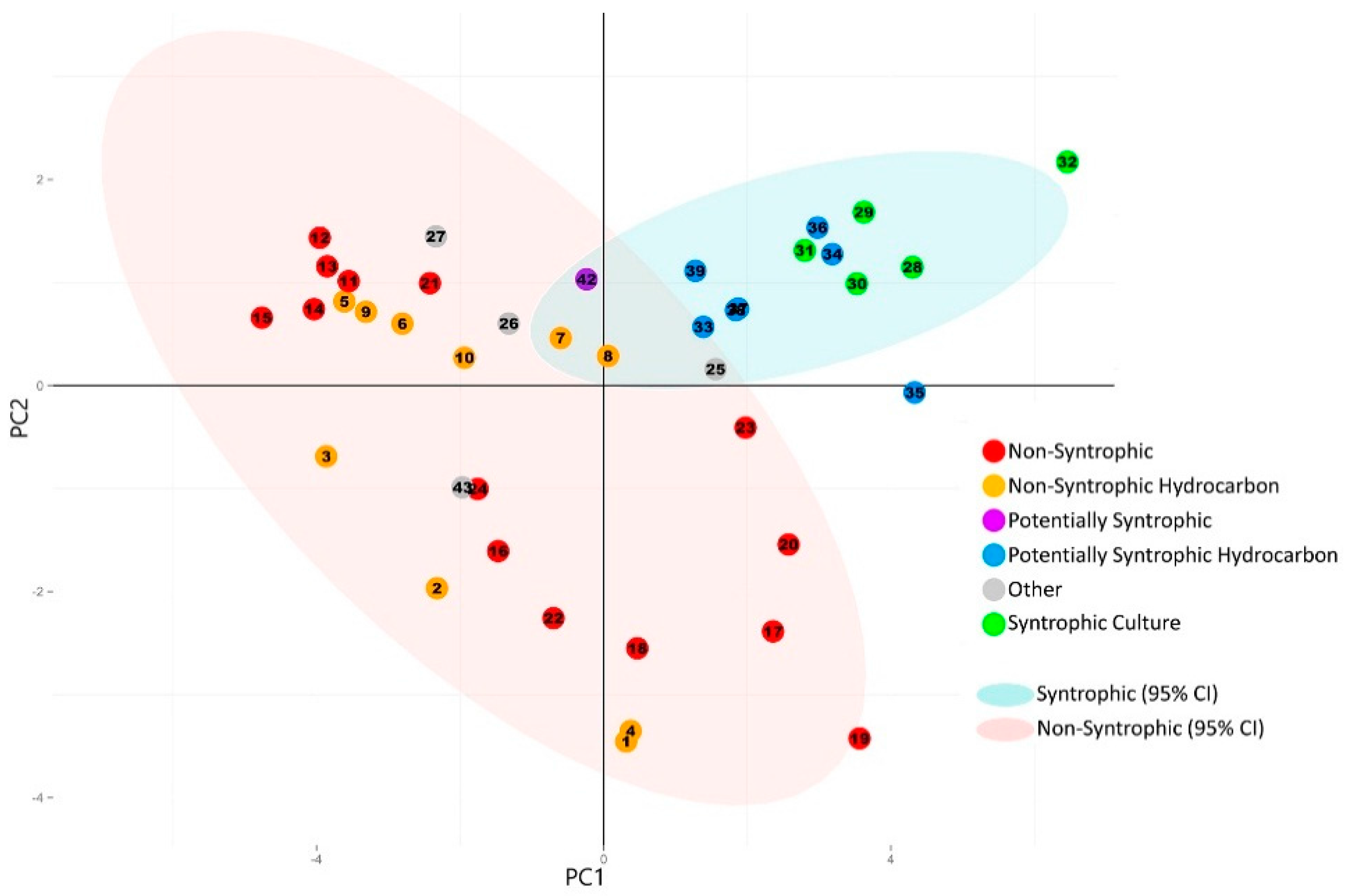
2.2. Metagenome Community Composition and Classification
2.3. Selection of Clusters of Orthologous Groups (COGs)
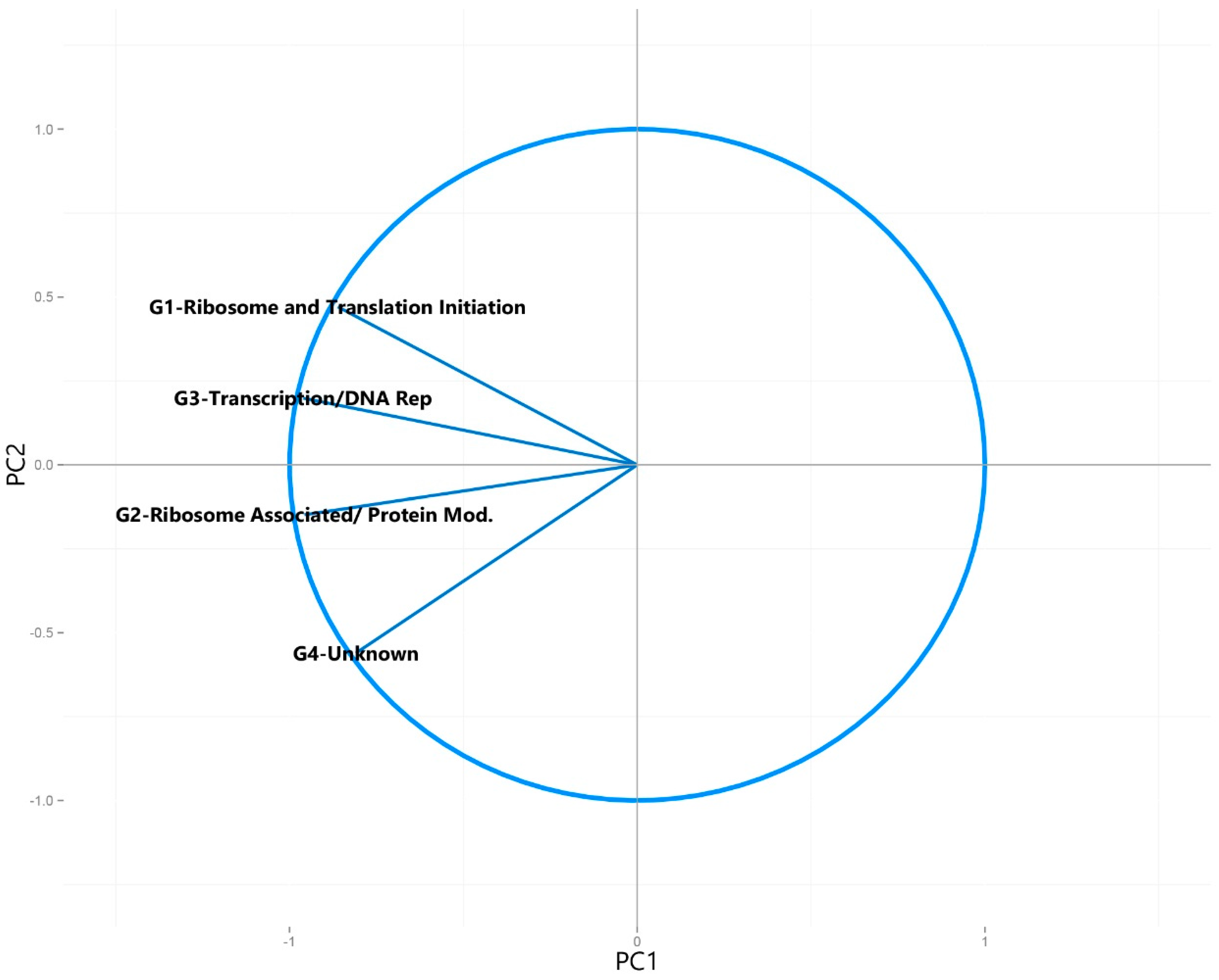
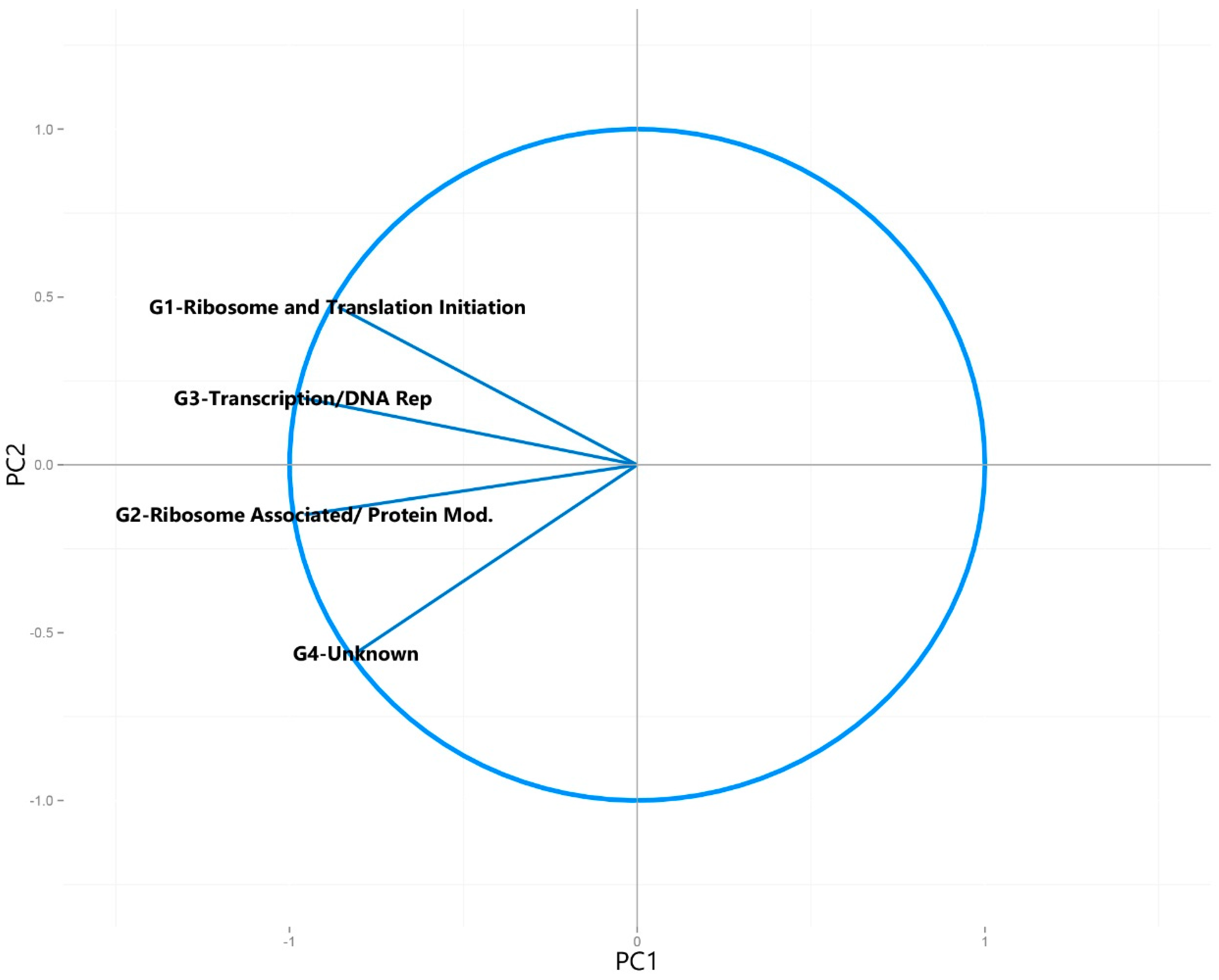
| Universally Present COG Categories | G1-Ribosome and Translation Initiation |
| G2-Ribosome Associated/ Protein Modification | |
| G3-Transcription/DNA Replication | |
| G4-Unknown | |
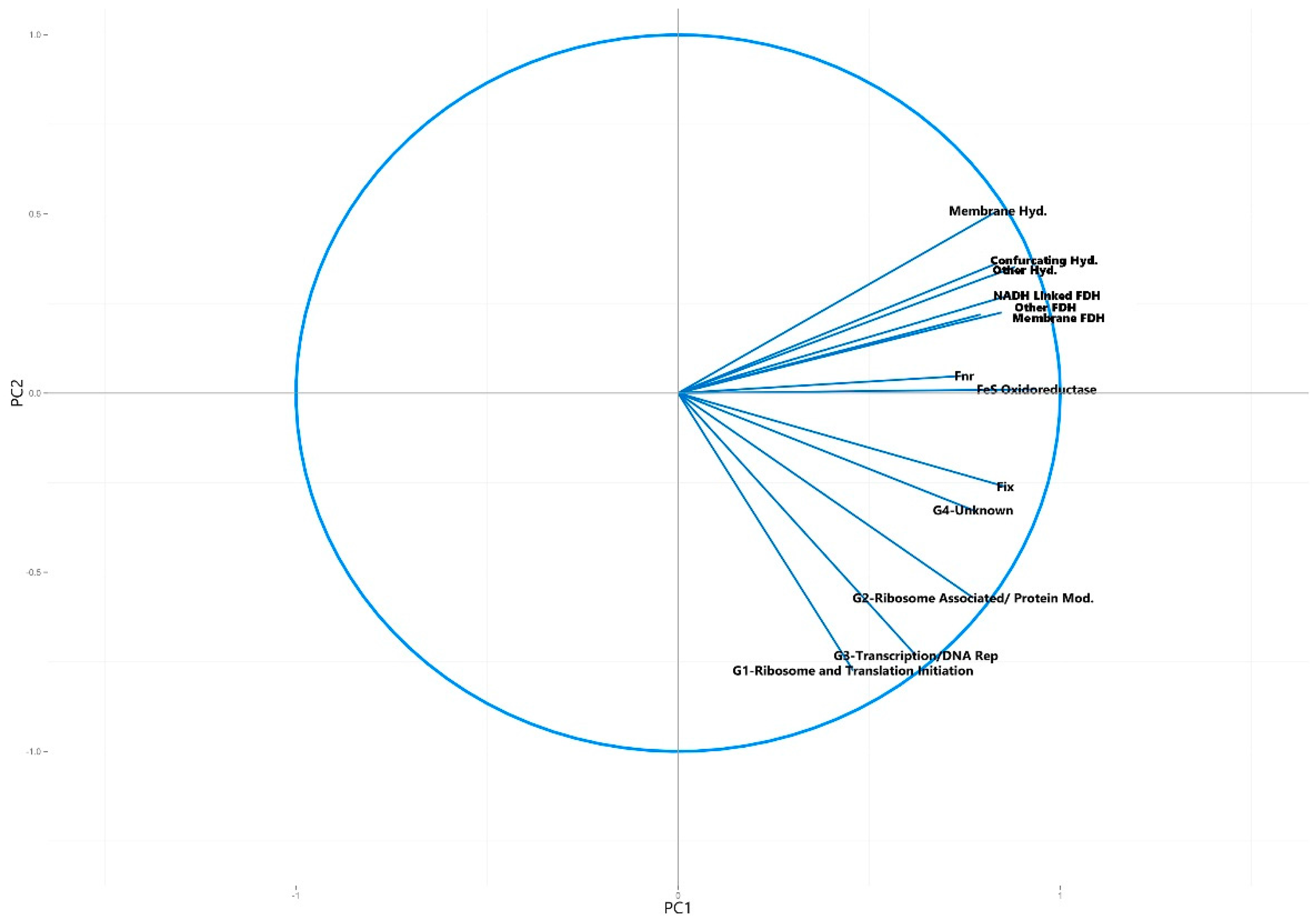
| Syntroph Associated COG Categories | FeS Oxidoreductases |
| Fnr | |
| Fix | |
| Confurcating Hydrogenases | |
| Other Hydrogenases | |
| Membrane Hydrogenases | |
| NADH Linked Formate Dehydrogenases | |
| Other Formate Dehydrogenases | |
| Membrane Formate Dehydrogenases |
2.4. Principal Component Analysis
2.5. Analysis of COGs in Metagenomes
3. Results
3.1. Metagenome Selection and Classification
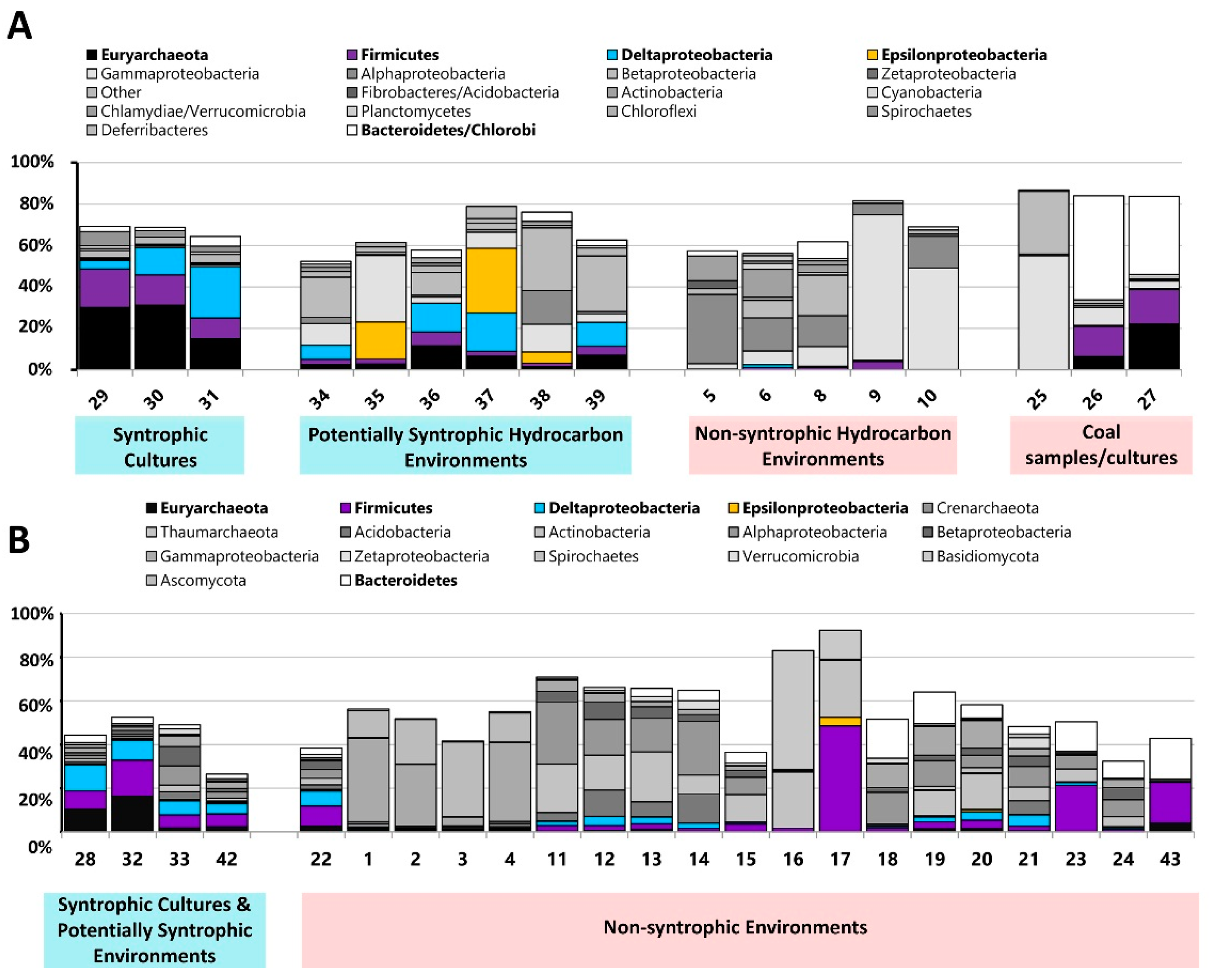
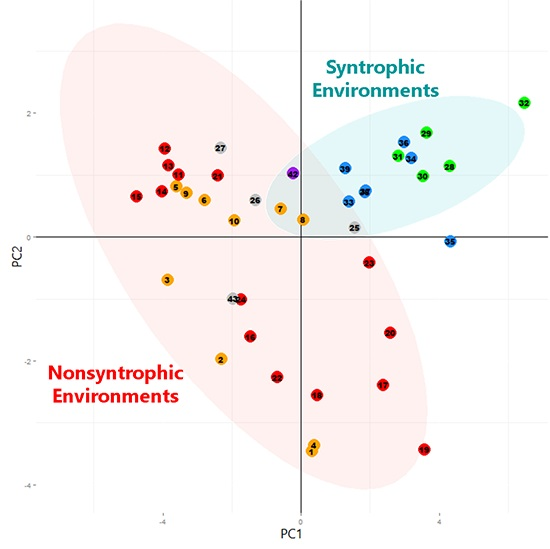
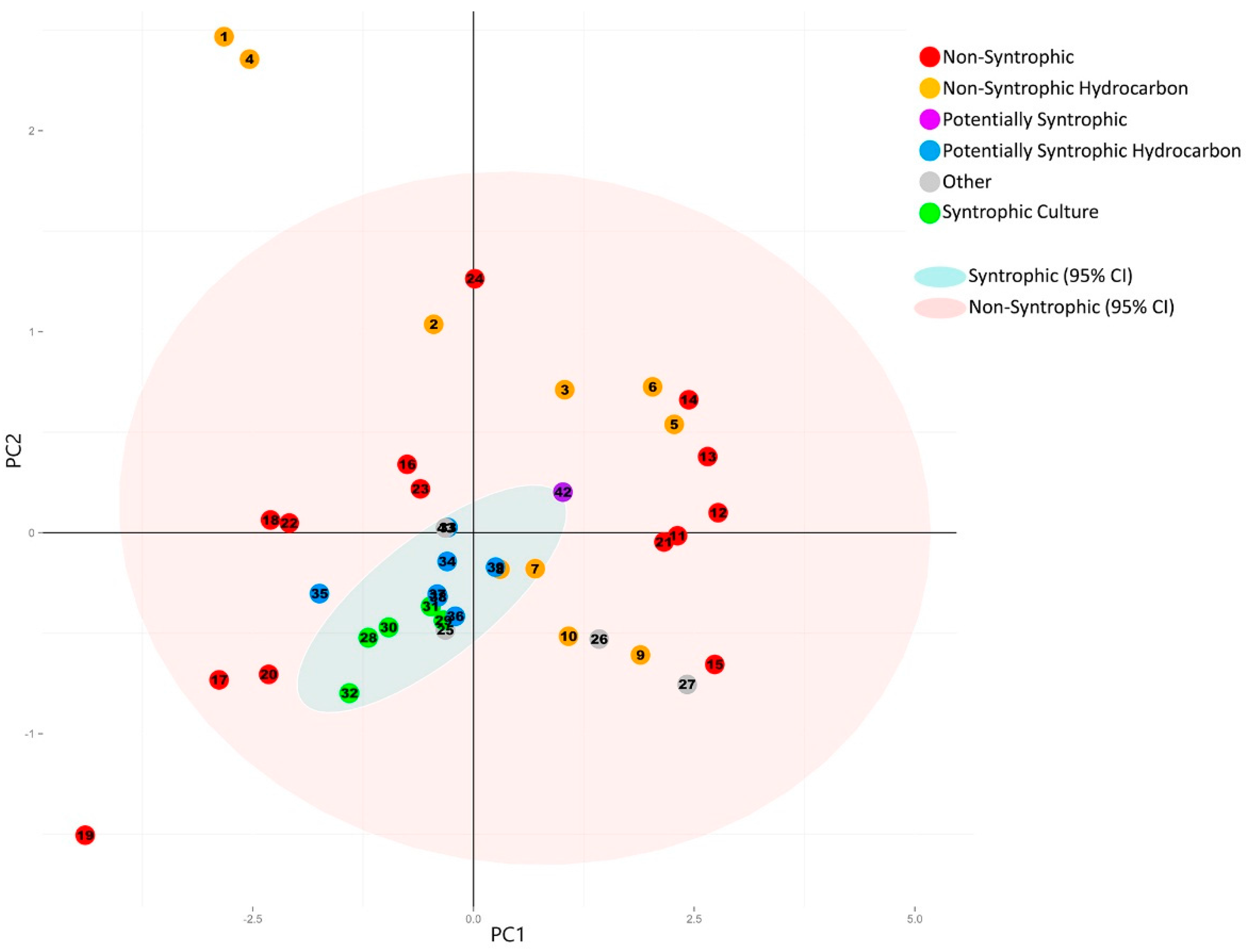
3.2. Principal Component Analysis
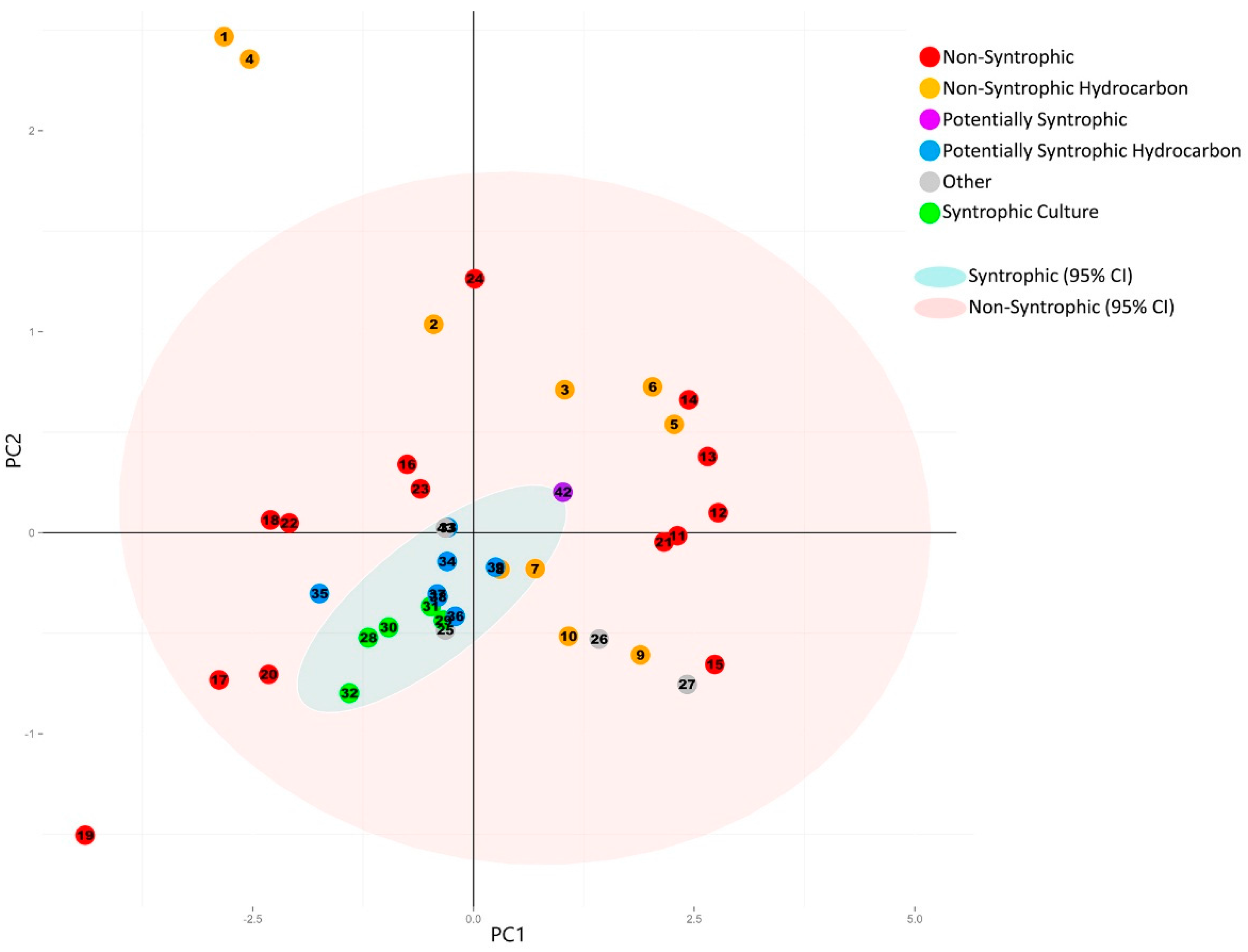
3.2.1. Principal Component Analysis of Universally Present COGs
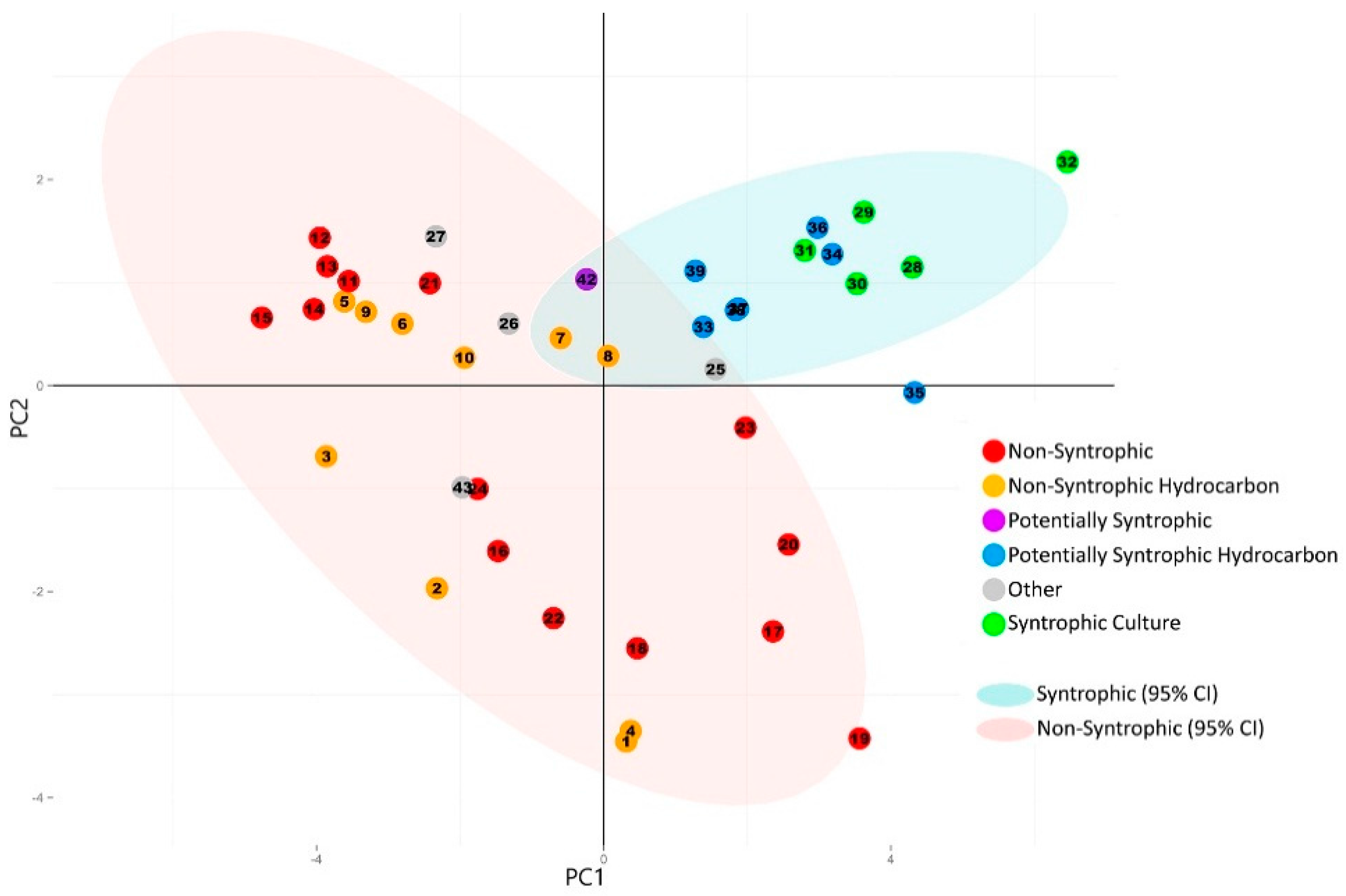
3.2.2. Principal Component Analysis of Syntroph-Associated and Universally Present COGs

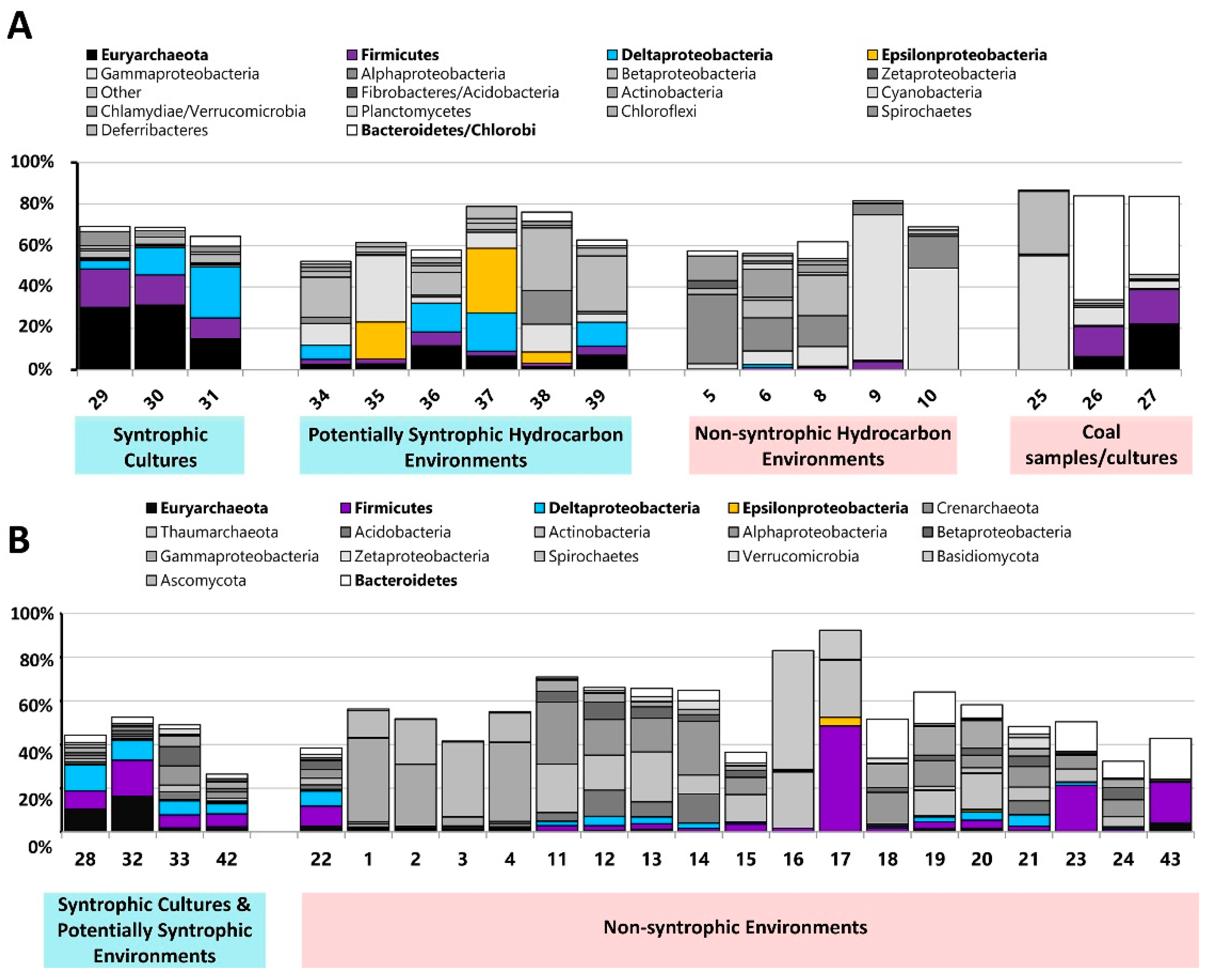
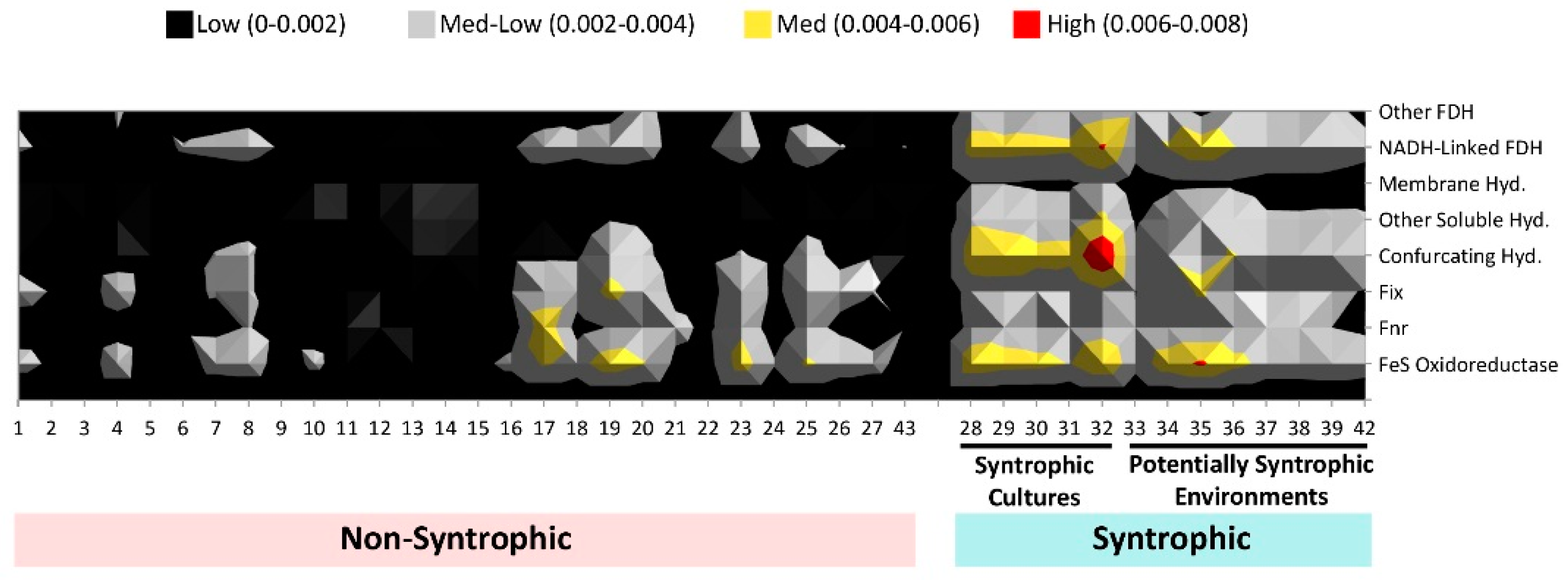
3.3. Comparison of Number of Syntroph-Associated COGs across Metagenomes
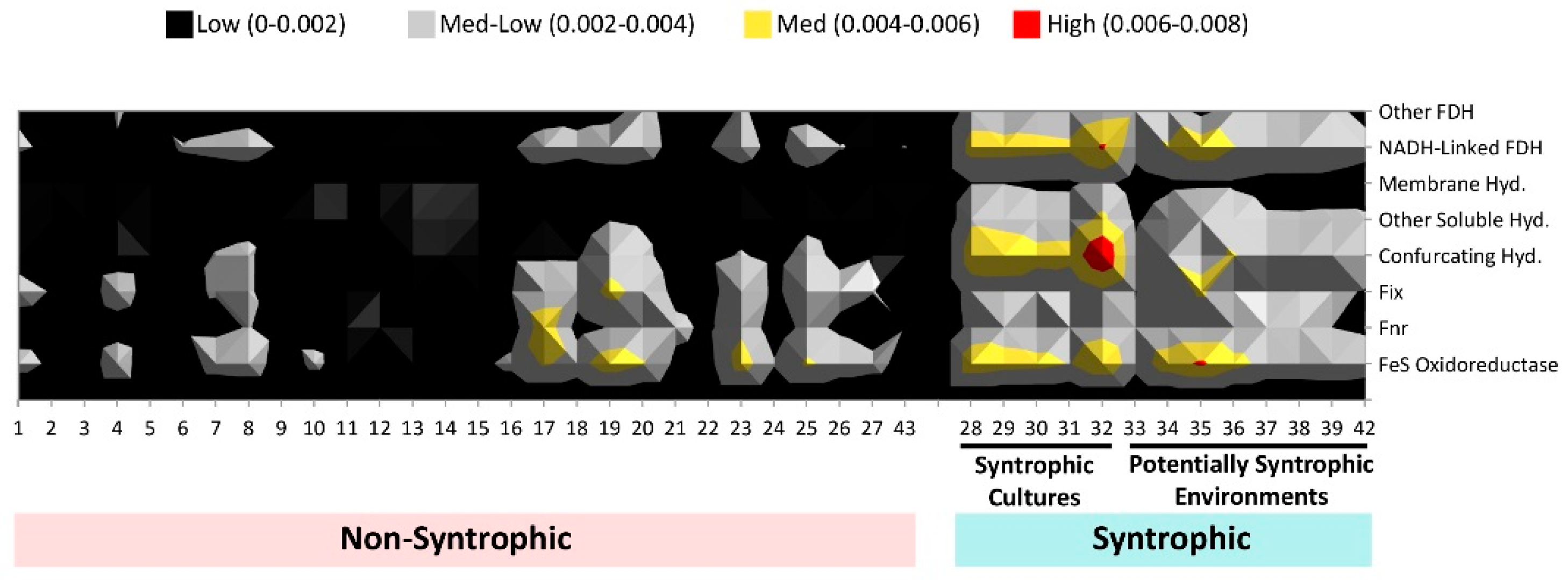
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Widdel, F.; Knittel, K.; Galushko, A. Anaerobic hydrocarbon-degrading microorganisms: An overview. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K.N., McGenity, T.J., van der Meer, J.R., de Lorenzo, V., Eds.; Springer-Verlag: Berlin, Germany, 2010; pp. 1997–2021. [Google Scholar]
- Agrawal, A.; Gieg, L.M. In situ detection of anaerobic alkane metabolites in subsurface environments. Front. Microbiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Gieg, L.M.; Fowler, S.J.; Berdugo-Clavijo, C. Syntrophic biodegradation of hydrocarbon contaminants. Curr. Opin. Biotechnol. 2014, 27, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Hubert, C.R.J.; Oldenburg, T.B.P.; Fustic, M.; Gray, N.D.; Larter, S.R.; Penn, K.; Rowan, A.K.; Seshadri, R.; Sherry, A.; Swainsbury, R.; et al. Massive dominance of Epsilonproteobacteria in formation waters from a Canadian oil sands reservoir containing severely biodegraded oil. Environ. Microbiol. 2012, 14, 387–404. [Google Scholar] [CrossRef] [PubMed]
- McInerney, M.J.; Sieber, J.R.; Gunsalus, R.P. Syntrophy in anaerobic global carbon cycles. Curr. Opin. Biotechnol. 2009, 20, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.E.L.; Henneberger, R.; Huber, H.; Moissl-Eichinger, C. Microbial syntrophy: Interaction for the common good. FEMS Microbiol. Rev. 2013, 37, 384–406. [Google Scholar] [CrossRef] [PubMed]
- Dolfing, J.; Larter, S.R.; Head, I.M. Thermodynamic constraints on methanogenic crude oil biodegradation. ISME J. 2008, 2, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Sieber, J.R.; McInerney, M.J.; Gunsalus, R.P. Genomic insights into syntrophy: The paradigm for anaerobic metabolic cooperation. Annu. Rev. Microbiol. 2012, 66, 429–452. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Caffrey, S.M.; Soh, J.; Agrawal, A.; Brown, D.; Budwill, K.; Dong, X.; Dunfield, P.F.; Foght, J.; Gieg, L.M.; et al. Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common. Environ. Sci. Technol. 2013, 47, 10708–10717. [Google Scholar] [CrossRef] [PubMed]
- The Hydrocarbon Metagenomics Project. Available online: http://www.hmp.ucalgary.ca (accessed on 29 October 2015).
- Summers, Z.M.; Fogarty, H.E.; Leang, C.; Franks, A.E.; Malvankar, N.S.; Lovley, D.R. Direct exchange of electrons within aggregates of an evolved syntrophic coculture of anaerobic bacteria. Science 2010, 330, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- De Bok, F.; Plugge, C.; Stams, A. Interspecies electron transfer in methanogenic propionate degrading consortia. Water Res. 2004, 38, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Edgren, T.; Nordlund, S. The fixABCX genes in Rhodospirillum rubrum encode a putative membrane complex participating in electron transfer to nitrogenase. J. Bacteriol. 2004, 186, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Biegel, E.; Müller, V. Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc. Natl. Acad. Sci. USA 2010, 107, 18138–18142. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Woyke, T.; Huntemann, M.; et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014, 42, D560–D567. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.M.A.; Chu, K.; Szeto, E.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Pagani, I.; Tringe, S.; et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 2014, 42, D568–D573. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.K.; Kelley, S.T.; Spiegelman, G.B.; Pace, N.R. The genetic core of the universal ancestor. Genome Res. 2003, 13, 407–412. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org (accessed on 29 October 2015).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer Science & Business Media: Medford, MA, USA, 2009. [Google Scholar]
- Fox, J.; Weisberg, S. The Car Package for R. An R Companion to Applied Regression, 2nd ed.; SAGE Publications: Thousand Oaks, CA, USA, 2011. [Google Scholar]
- Wickham, H.; Winston, C. Devtools: Tools to Make Developing R Packages Easier. Available online: https://cran.r-project.org/package=devtools (accessed on 29 October 2015).
- Eddelbuettel, D.; Lucas, A.; Tuszynski, J.; Bengtsson, H.; Urbanek, S.; Frasca, M.; Lewis, B.; Stokely, M.; Muehleisen, H.; Murdoch, D. Digest: Create Cryptographic Hash Digests of R Objects. Available online: http://finzi.psych.upenn.edu/library/digest/html/00Index.html (accessed on 29 October 2015).
- Strapoc, D.; Picardal, F.W.; Turich, C.; Schaperdoth, I.; Macalady, J.L.; Lipp, J.S.; Lin, Y.S.; Ertefai, T.F.; Schubotz, F.; Hinrichs, K.U.; et al. Methane-producing microbial community in a coal bed of the Illinois basin. Appl. Environ. Microbiol. 2008, 74, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Sonnenburg, J.L. Eating for two: How metabolism establishes interspecies interactions in the gut. Cell Host Microbe 2011, 10, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Qiu, Q.; Liu, P.; Rui, J.; Lu, Y. Syntrophic oxidation of propionate in rice field soil at 15 and 30 °C under methanogenic conditions. Appl. Environ. Microbiol. 2012, 78, 4923–4932. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.S.; Thahira-Rahman, J.; Lakshmanaperumalsamy, P.; Banat, I. Towards efficient crude oil degradation by a mixed bacterial consortium. Bioresour. Technol. 2002, 85, 257–261. [Google Scholar] [CrossRef]
- Jones, E.J.P.; Voytek, M.A.; Corum, M.D.; Orem, W.H. Stimulation of methane generation from nonproductive coal by addition of nutrients or a microbial consortium. Appl. Environ. Microbiol. 2010, 76, 7013–7022. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oberding, L.; Gieg, L.M. Metagenomic Analyses Reveal That Energy Transfer Gene Abundances Can Predict the Syntrophic Potential of Environmental Microbial Communities. Microorganisms 2016, 4, 5. https://doi.org/10.3390/microorganisms4010005
Oberding L, Gieg LM. Metagenomic Analyses Reveal That Energy Transfer Gene Abundances Can Predict the Syntrophic Potential of Environmental Microbial Communities. Microorganisms. 2016; 4(1):5. https://doi.org/10.3390/microorganisms4010005
Chicago/Turabian StyleOberding, Lisa, and Lisa M. Gieg. 2016. "Metagenomic Analyses Reveal That Energy Transfer Gene Abundances Can Predict the Syntrophic Potential of Environmental Microbial Communities" Microorganisms 4, no. 1: 5. https://doi.org/10.3390/microorganisms4010005
APA StyleOberding, L., & Gieg, L. M. (2016). Metagenomic Analyses Reveal That Energy Transfer Gene Abundances Can Predict the Syntrophic Potential of Environmental Microbial Communities. Microorganisms, 4(1), 5. https://doi.org/10.3390/microorganisms4010005