
Experimental Horizontal Gene Transfer of Methylamine Dehydrogenase Mimics Prevalent Exchange in Nature and Overcomes the Methylamine Growth Constraints Posed by the Sub-Optimal N-Methylglutamate Pathway

Abstract
:1. Introduction
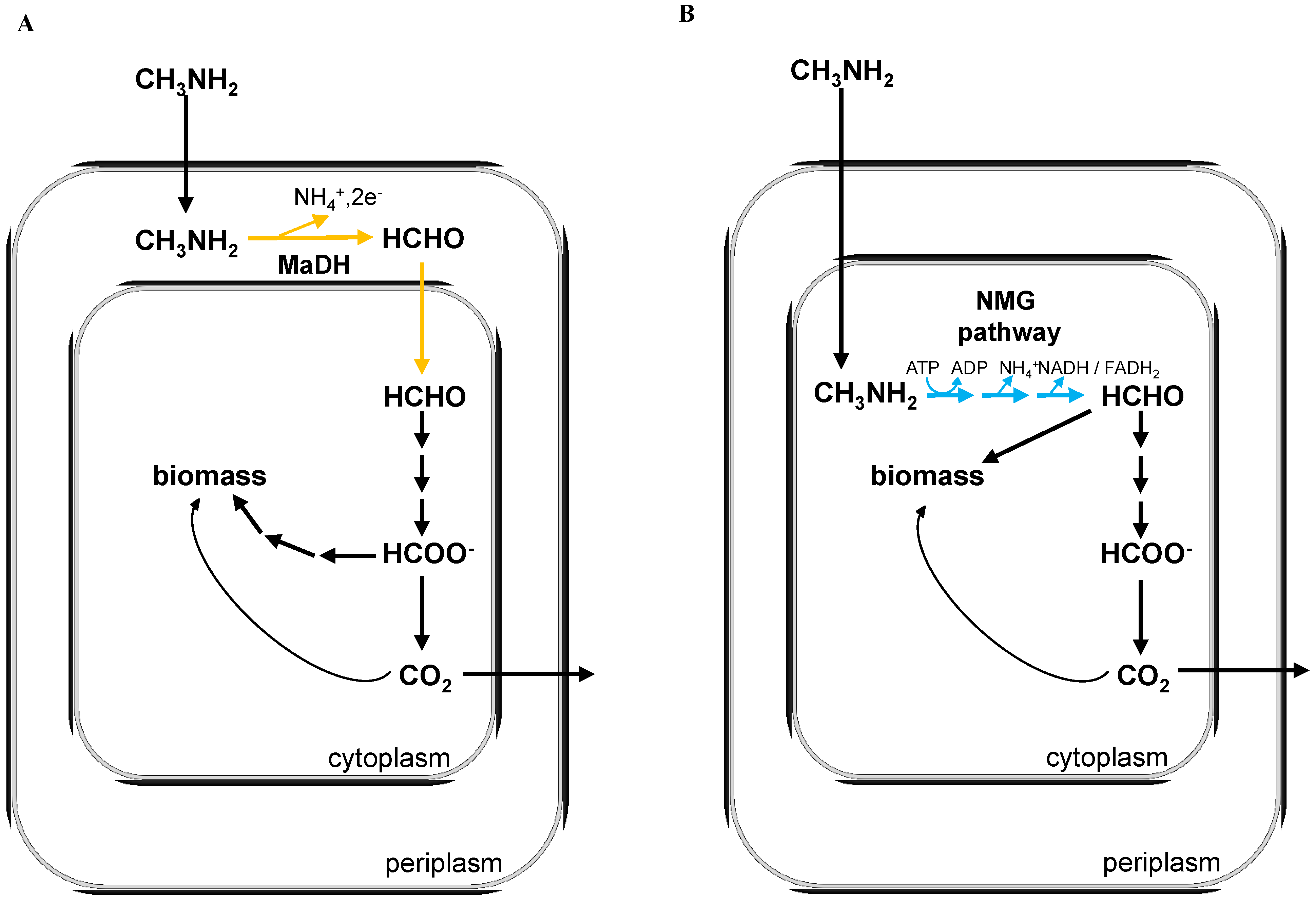
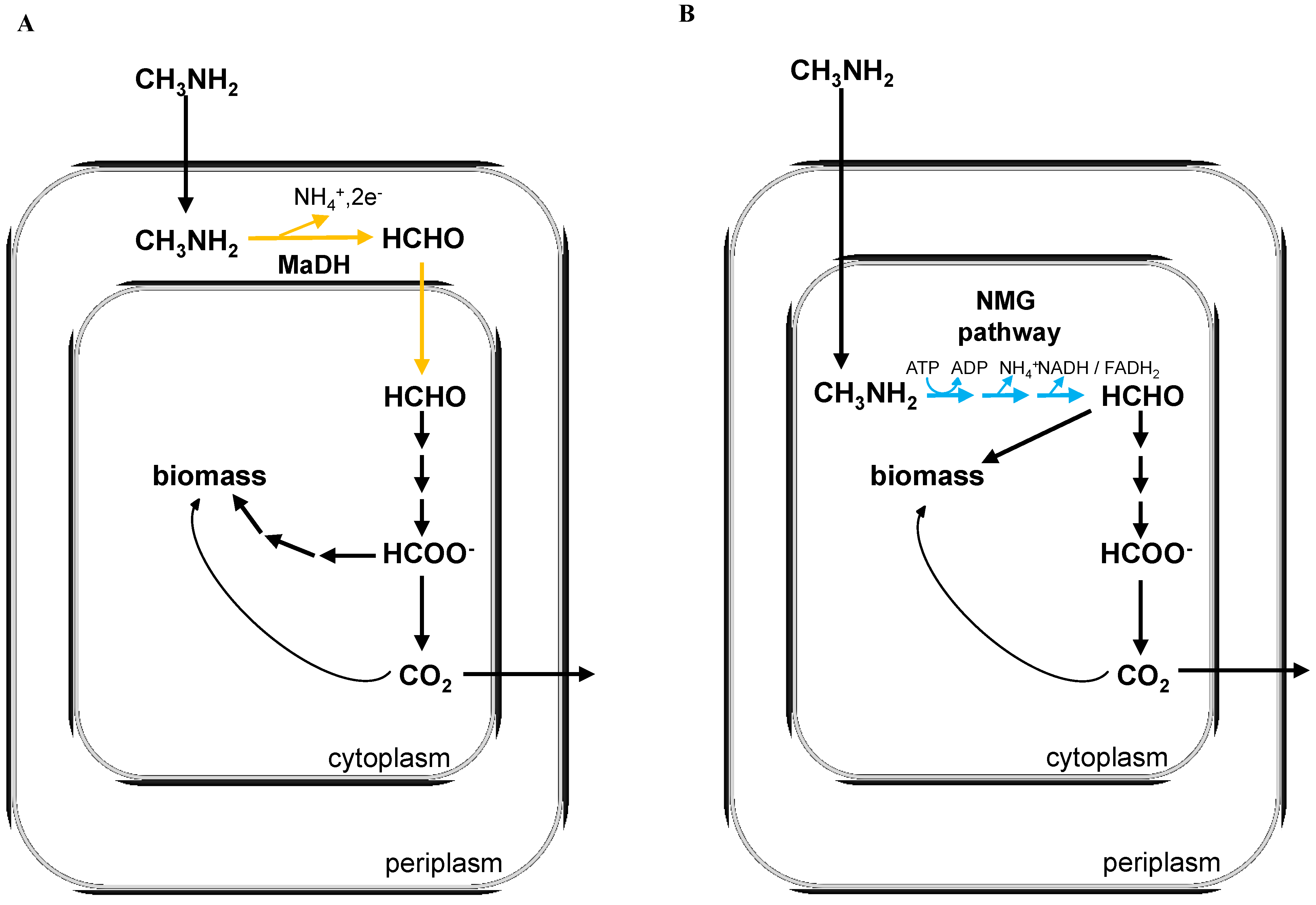
2. Experimental Section
2.1. Chemicals and Media
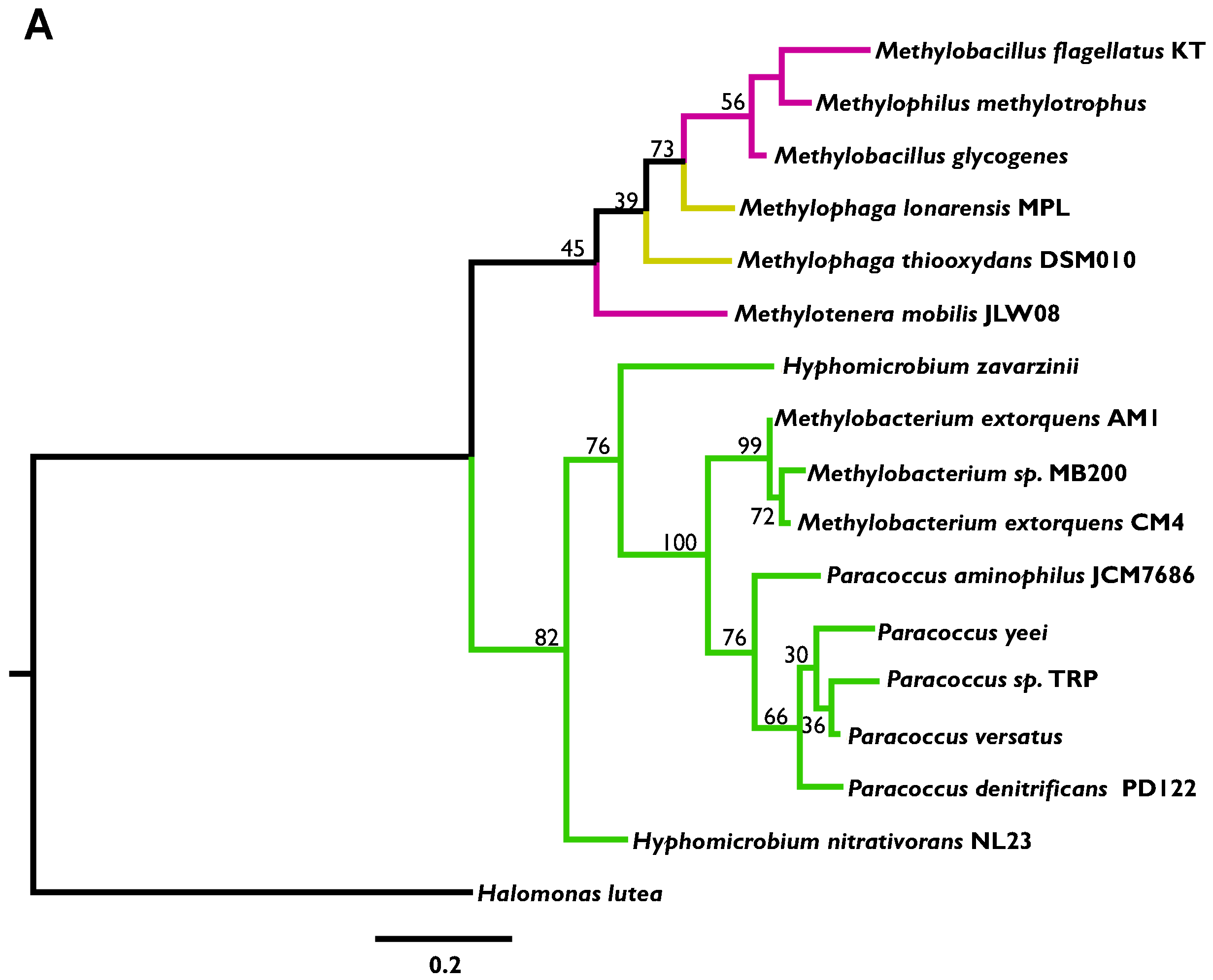
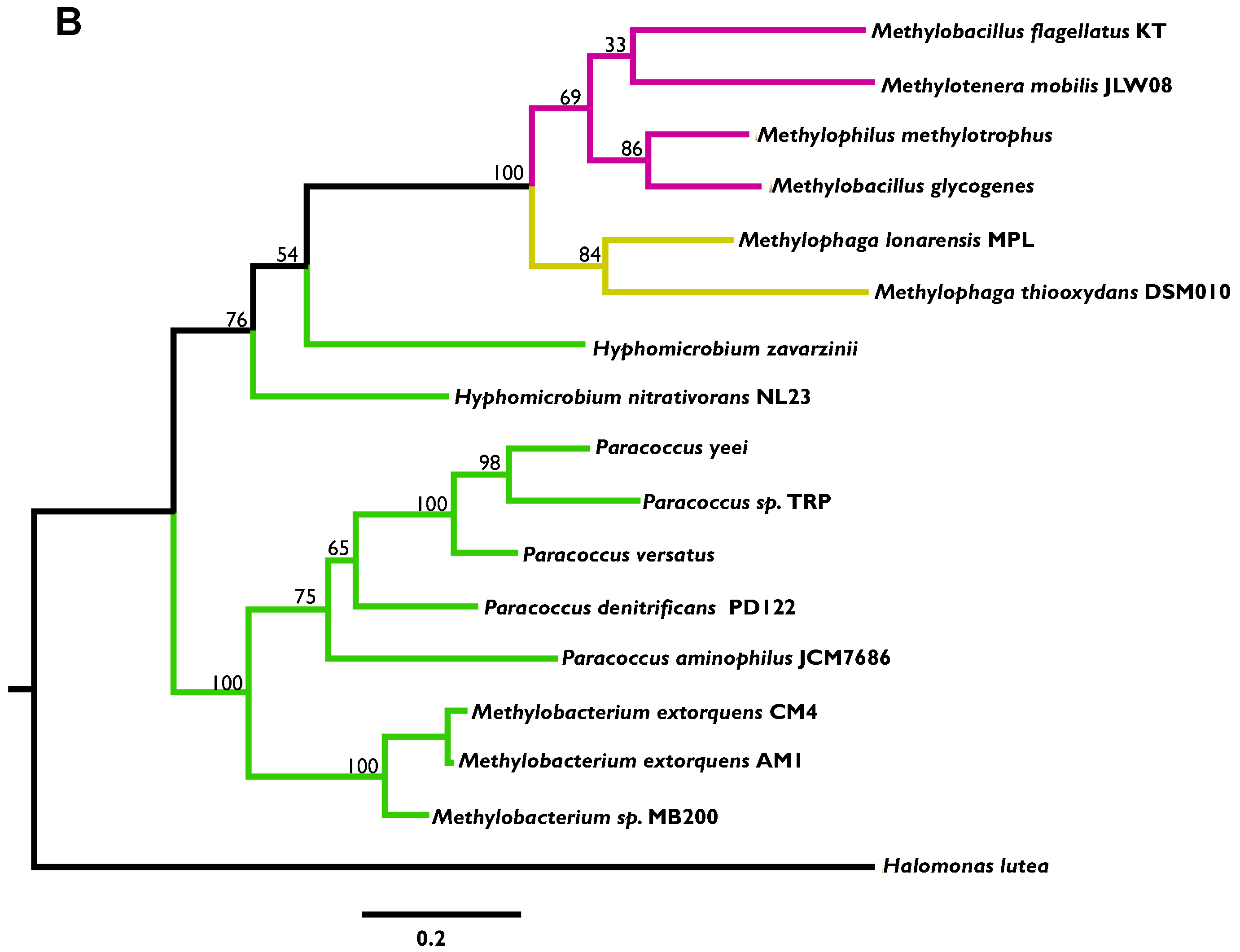
2.2. Phylogenetic Analysis
2.3. Experimental Evolution
2.4. Fitness Assays

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
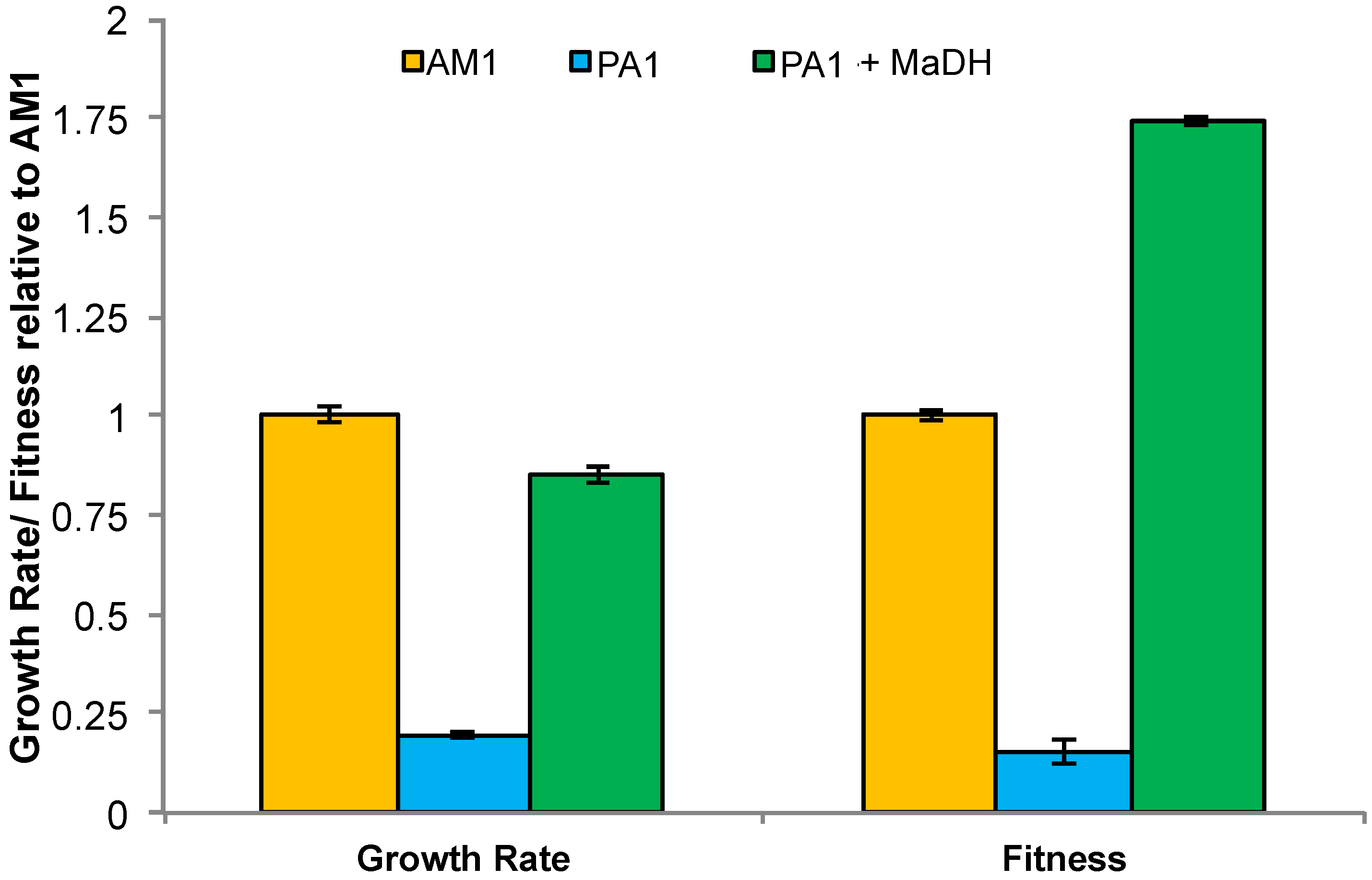{kind=link}
{kind=link}
| Strains or Plasmid | Description | Reference |
|---|---|---|
| Strains | ||
| CM4 | M. extorquens CM4 | [52] |
| DM4 | M. extorquens DM4 | [53] |
| BJ001 | M. extorquens BJ001 | [54] |
| CM2720 | ∆cel M. extorquens AM1 | [55] |
| CM2730 | ∆cel M. extorquens PA1 | [56] |
| CM3120 | ∆katA::Ptac-mCherry in CM2720a | [49] |
| CM3839 | ∆hpt::Ptac-mCherry in CM2730b | [49] |
| CM4408 | E1 | This study |
| CM4409 | E2 | This study |
| CM4410 | E3 | This study |
| Plasmids | ||
| pAYC139 | Plasmid (IncP, tra, TetR) with mau gene cluster from M. extorquens AM1 | [14] |
2.5. Growth Rate Measurement
3. Results
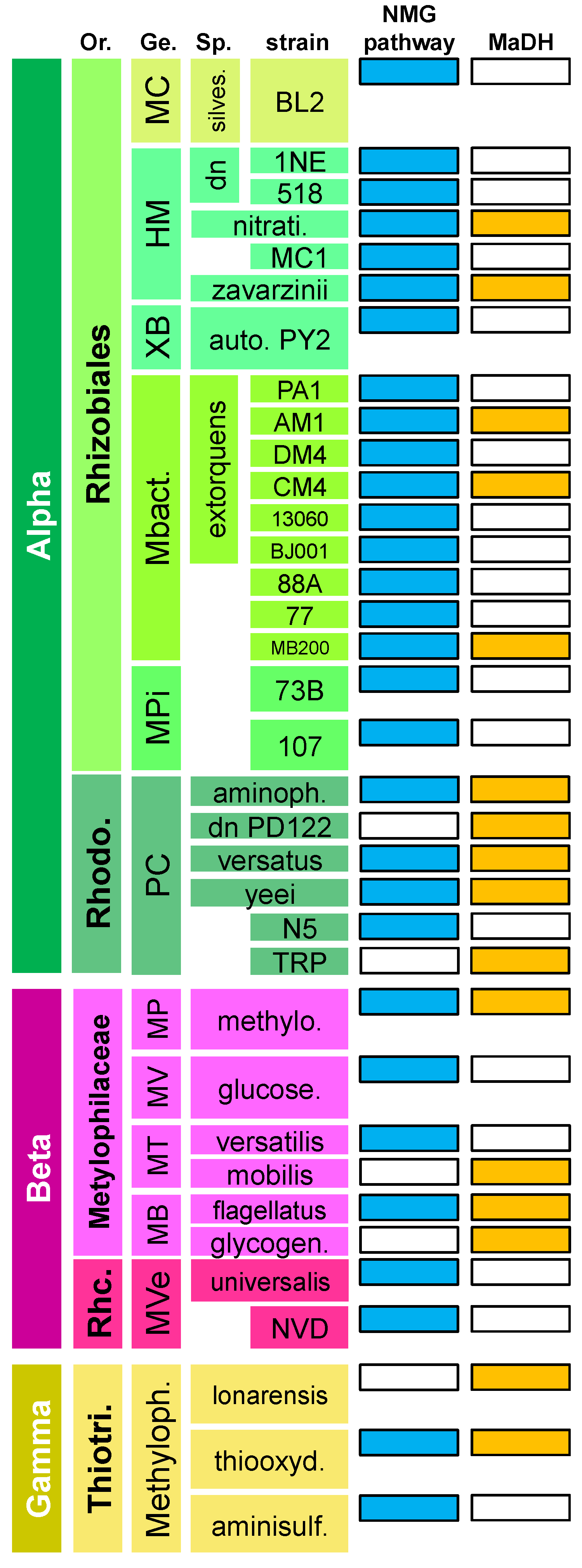
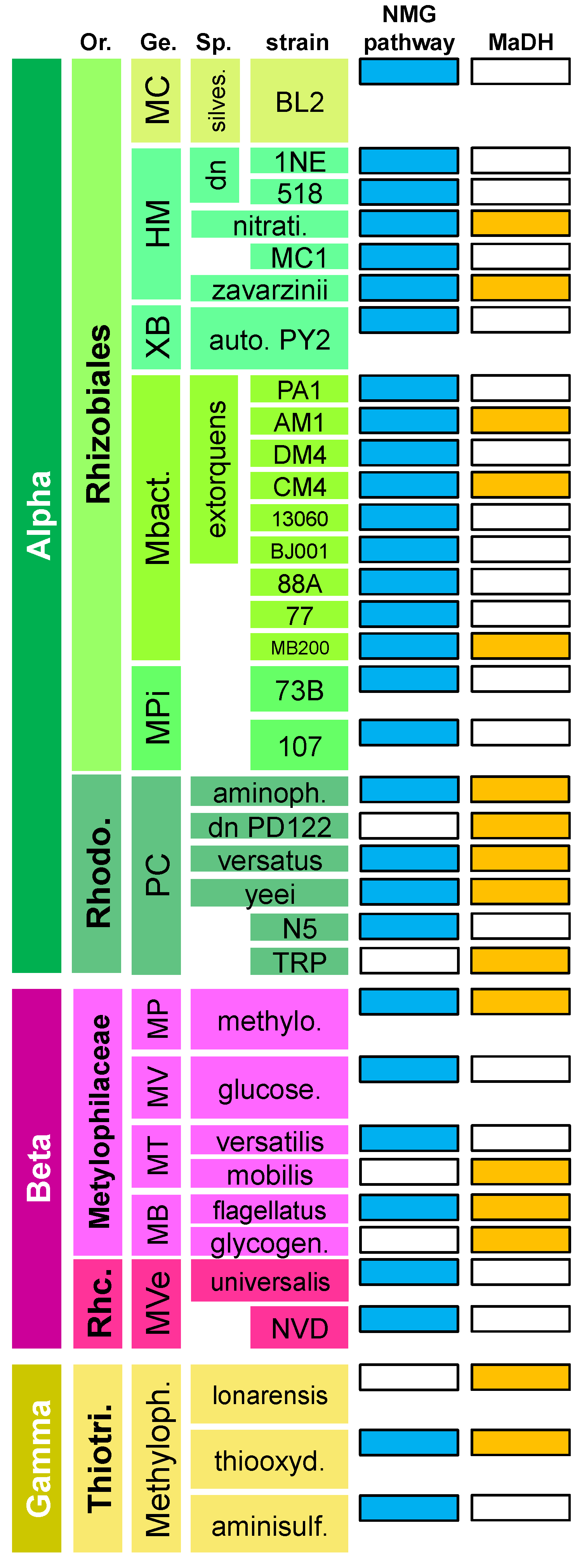
3.1. The N-Methylglutamate Pathway for Methylamine Oxidation is More Widely Distributed in Methylotrophic Proteobacteria than Methylamine Dehydrogenase
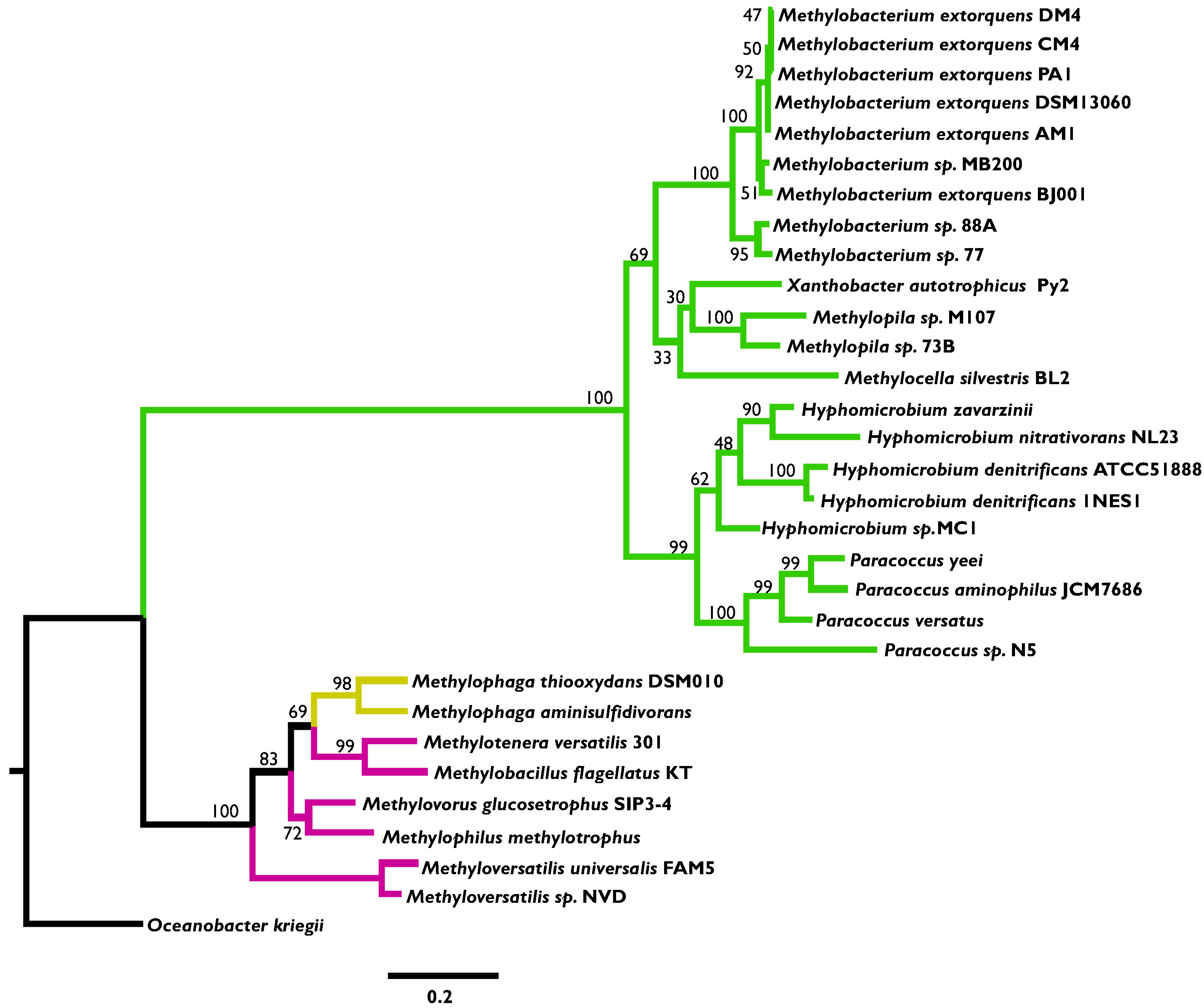
3.2. Phylogenetic Analysis Indicates More HGT for MaDH than the NMG Pathway
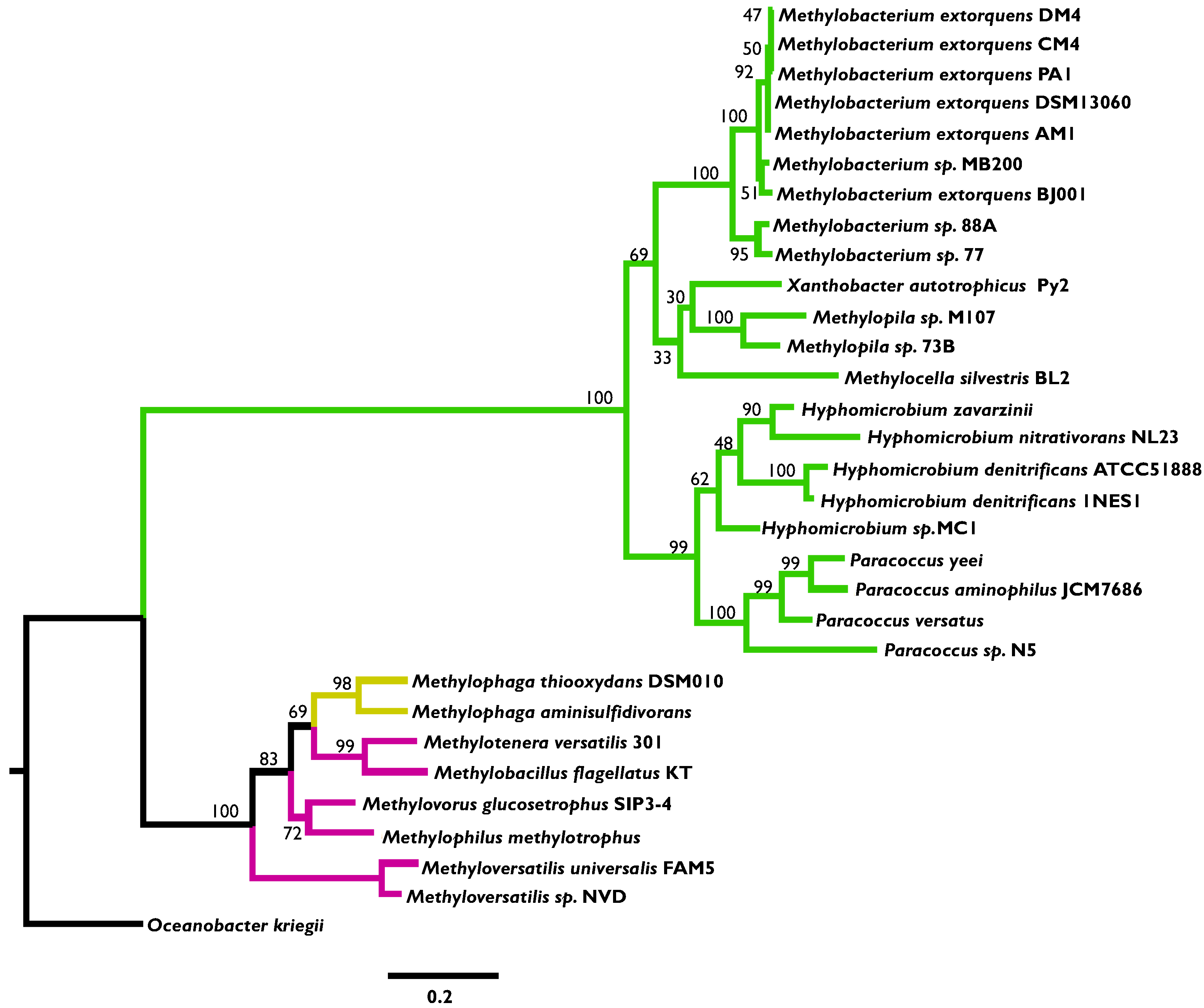
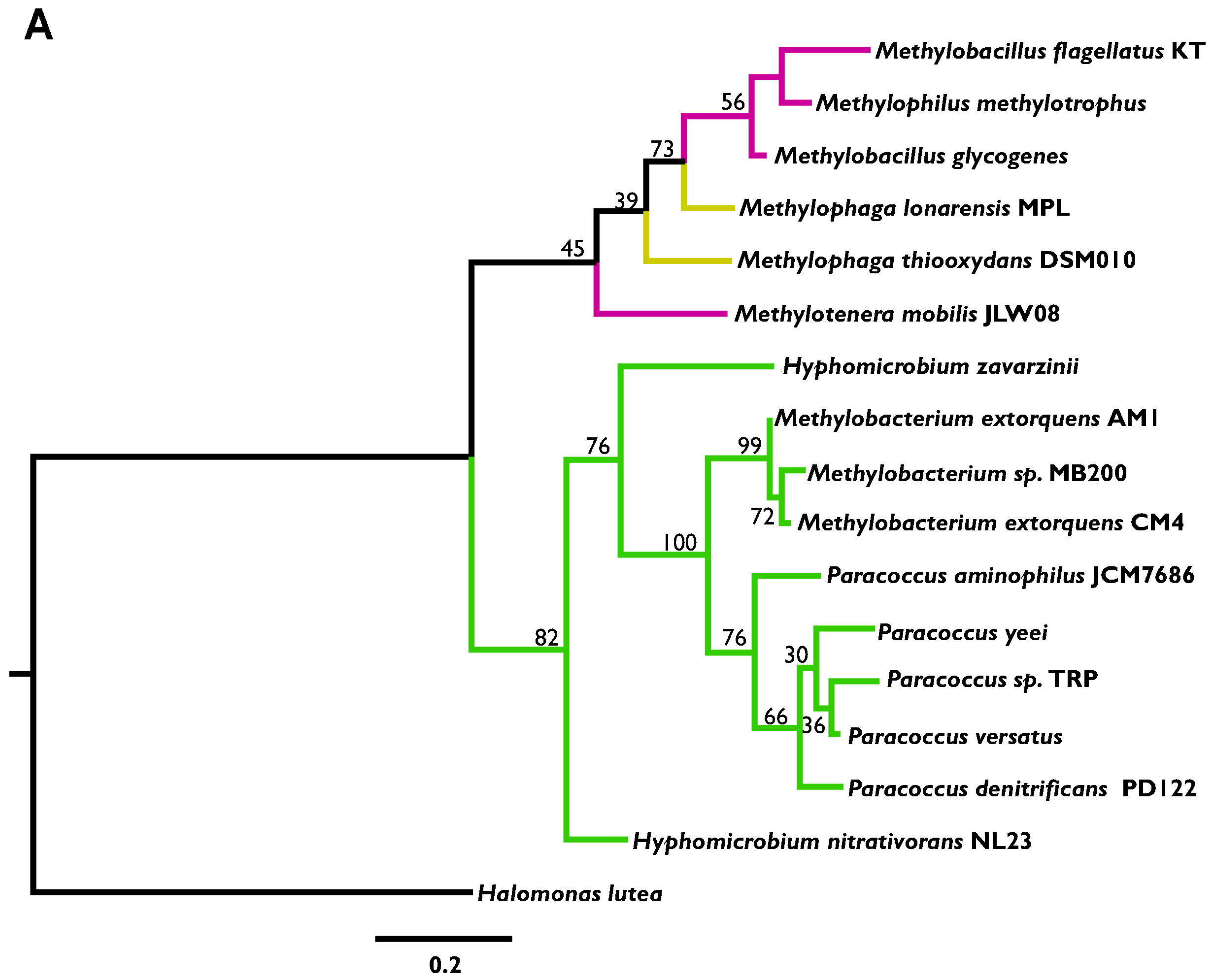
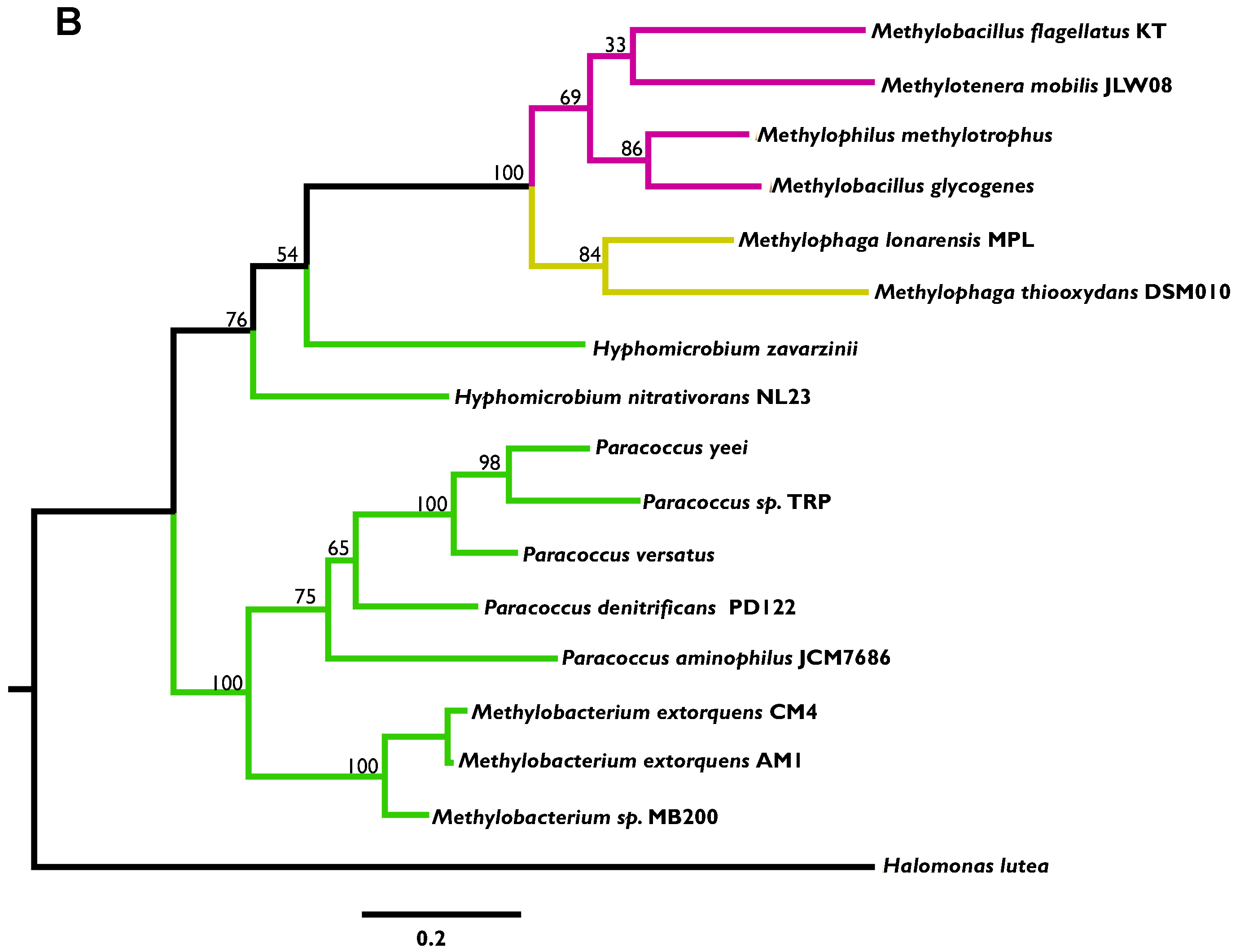
3.3. Highly Distinct Modes of Inheritance within Methylobacterium for Two Methylamine Oxidation Pathways
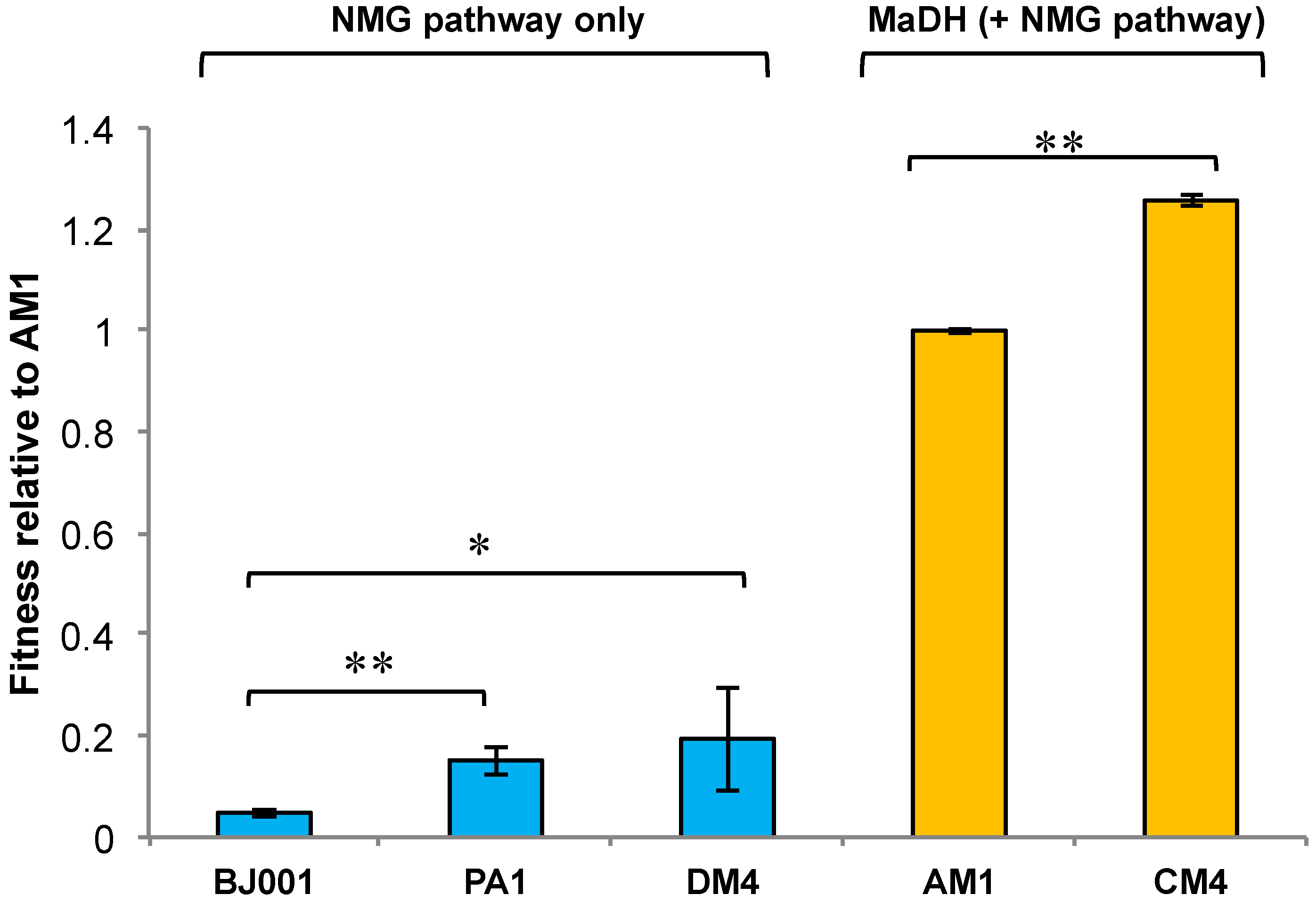
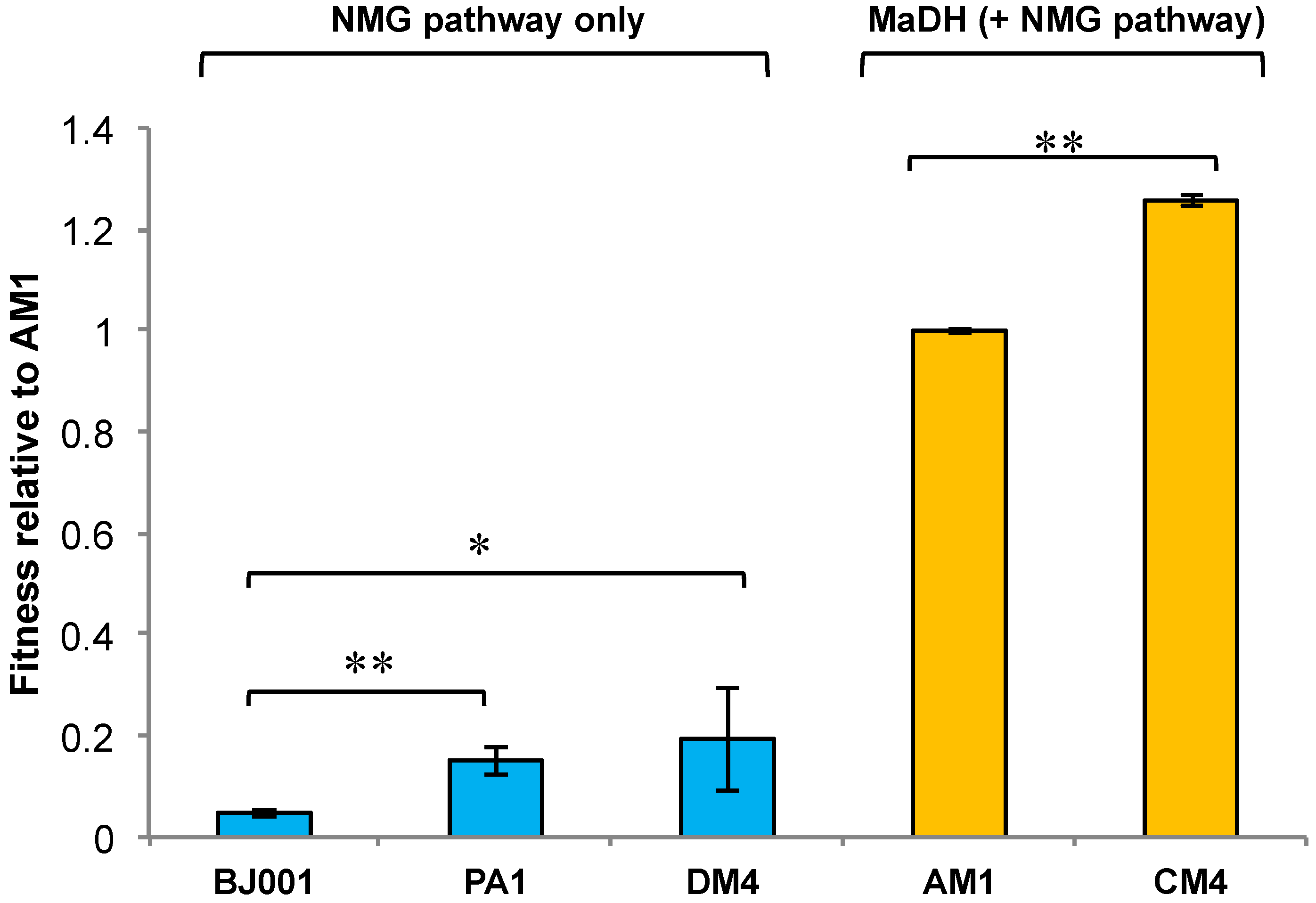
3.4. The Methylamine Fitness of M. extorquens Species Is Dependent on the Metabolic Module Used for Methylamine Oxidation
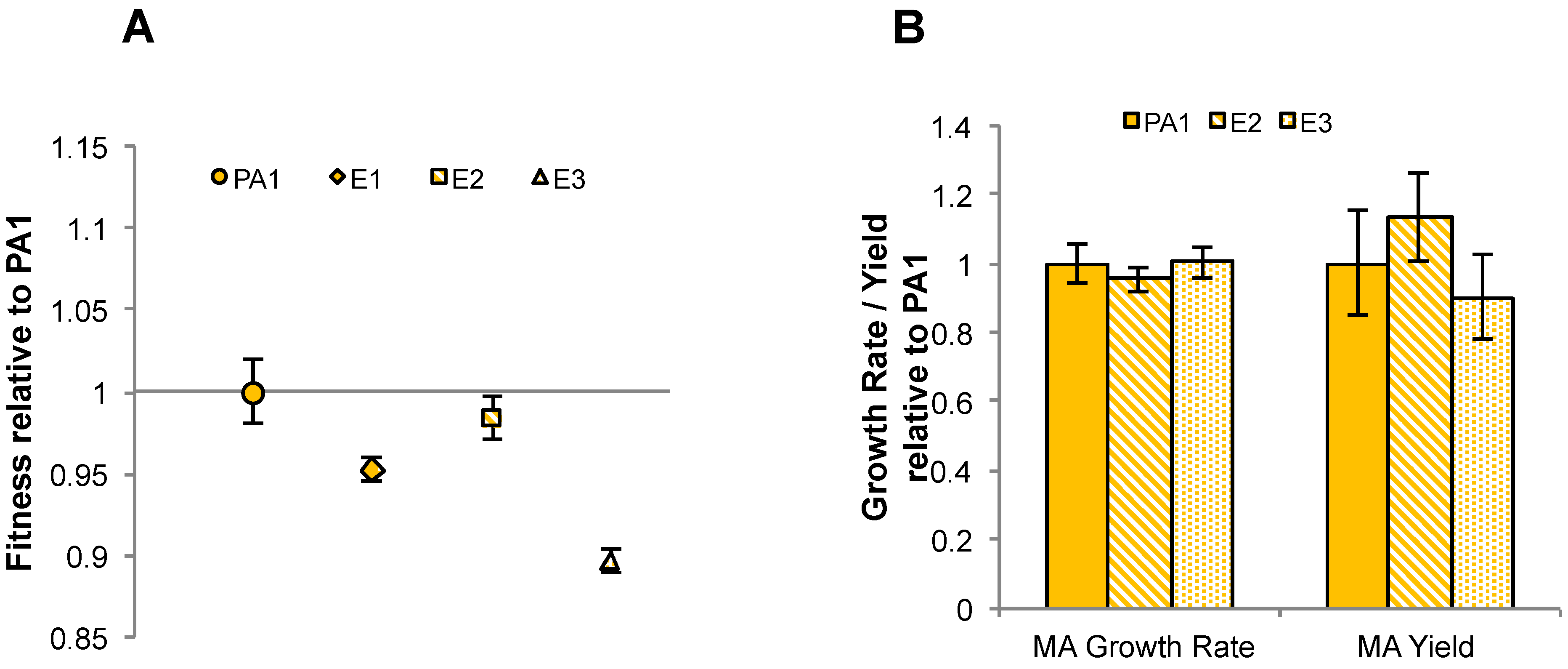
3.5. Experimental HGT of the mau Gene Cluster in M. extorquens PA1 Leads to an Instantaneous Increase in Fitness and Growth Rate on Methylamine
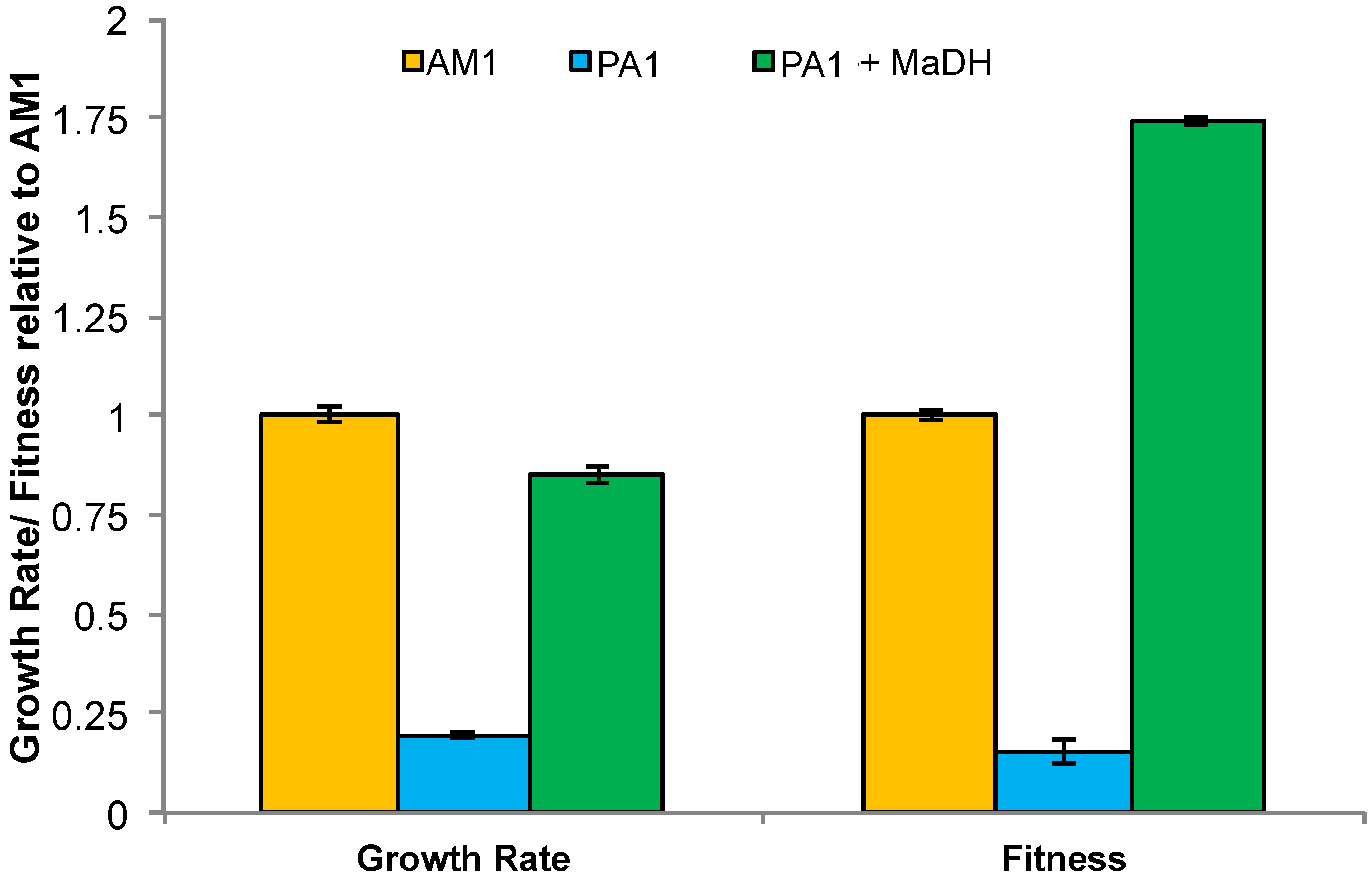
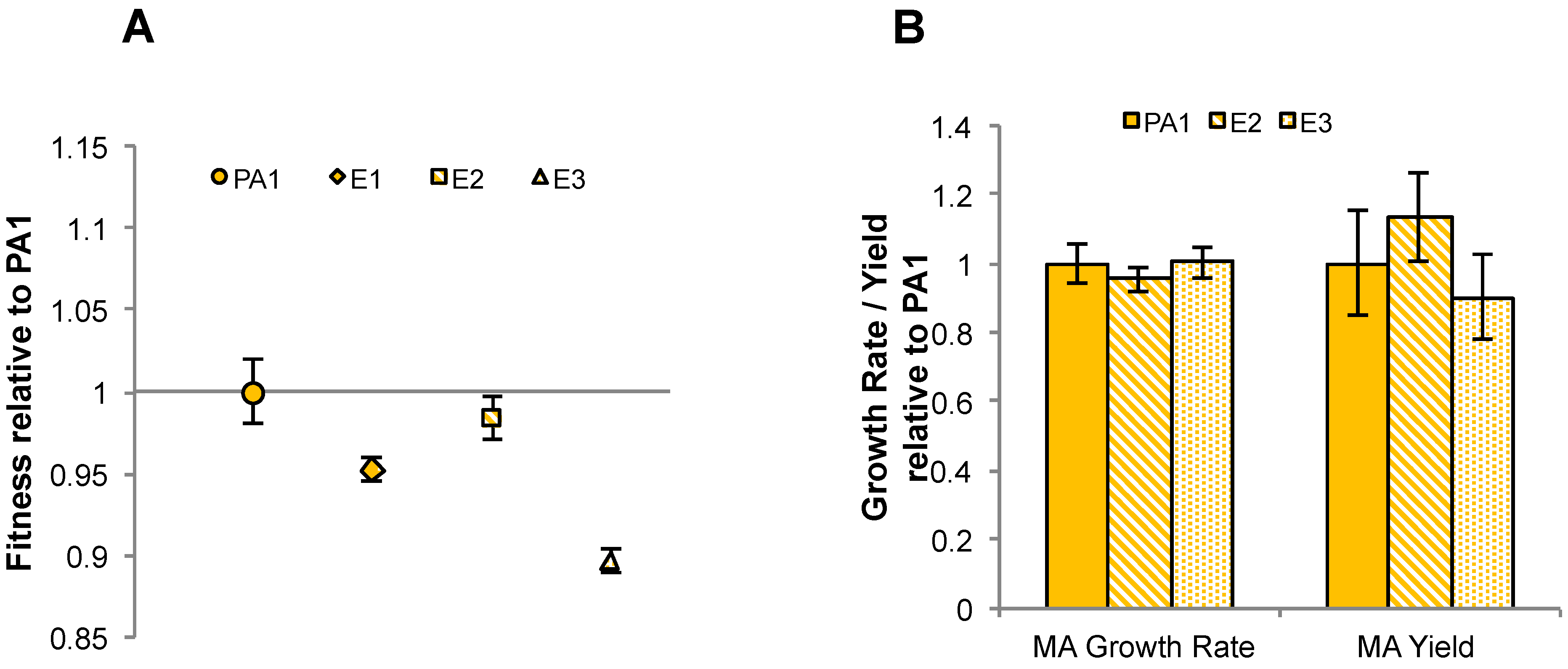
3.6. Laboratory-Based Evolution Did Not Lead to a Rapid Increase in Methylamine Fitness or Growth Rate for M. extorquens PA1
4. Discussion
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berman, T.; Bronk, D.A. Dissolved organic nitrogen: A dynamic participant in aquatic ecosystems. Aquat. Microb. Ecol. 2003, 31, 279–305. [Google Scholar] [CrossRef]
- Naqvi, S.W.A.; Bange, H.W.; Gibb, S.W.; Goyet, C.; Hatton, A.D.; Upstill-Goddard, R.C. Biogeochemical ocean-atmosphere transfers in the Arabian Sea. Prog. Oceanogr. 2005, 65, 116–144. [Google Scholar] [CrossRef]
- Fitzsimons, M.F.; Millward, G.E.; Revitt, D.M.; Dawit, M.D. Desorption kinetics of ammonium and methylamines from estuarine sediments: Consequences for the cycling of nitrogen. Mar. Chem. 2006, 101, 12–26. [Google Scholar] [CrossRef]
- Neufeld, J.D.; Schäfer, H.; Cox, M.J.; Boden, R.; McDonald, I.R.; Murrell, J.C. Stable-isotope probing implicates Methylophaga spp and novel Gammaproteobacteria in marine methanol and methylamine metabolism. ISME J. 2007, 1, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Worsfold, P.J.; Monbet, P.; Tappin, A.D.; Fitzsimons, M.F.; Stiles, D.A.; McKelvie, I.D. Characterization and quantification of organic phosphorous and organic nitrogen components in aquatic systems: A review. Anal. Chim. Acta 2008, 624, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Wexler, A.S.; Clegg, S.L. Atmospheric amines—Part 1. A review. Atmos. Env. 2011, 45, 524–546. [Google Scholar] [CrossRef]
- Wischer, A.; Kumaresan, D.; Johnston, A.; El Khawand, M.; Stephenson, J.; Hillebrand-Voiculescu, A.M.; Chen, Y.; Murrell, J.C. Bacterial metabolism of methylated amines and identification of novel methylotrophs in Movile Cave. ISME J. 2015, 9, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Lovely, D.R.; Klug, M.J. Methanogenesis from methanol and methylamines and acetogenesis from hydrogen and carbon dioxide in the sediments of a eutrophic lake. Appl. Environ. Microbiol. 1983, 45, 1310–1315. [Google Scholar] [PubMed]
- Cicerone, R.J.; Oremland, R.S. Biogeochemical aspects of atmospheric methane. Glob. Biogeochem. Cycles 1988, 2, 299–327. [Google Scholar] [CrossRef]
- Oren, A. Formation and breakdown of glycine betaine and trimethylamine in hypersaline environments. Antonie Leeuwenhoek 1990, 58, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Anthony, C. The Biochemistry of Methylotrophs; Academic Press Ltd.: London, UK, 1982. [Google Scholar]
- Chistoserdova, L.; Kalyuzhnaya, M.G.; Lidstrom, M.E. The expanding world of methylotrophic metabolism. Annu. Rev. Microbiol. 2009, 63, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L. Modularity of methylotrophy, revisited. Environ. Microbiol. 2011, 13, 2603–2622. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdov, A.Y.; Tsygankov, Y.D.; Lidstrom, M.E. Genetic organization of methylamine utilization genes from Methylobacterium extorquens AM1. J. Bacteriol. 1991, 173, 5901–5908. [Google Scholar] [PubMed]
- Latypova, E.; Yang, S.; Wang, Y.S.; Wang, T.; Chavkin, T.A.; Hackett, M.; Schäfer, H.; Kalyuzhnaya, M.G. Genetics of the glutamate-mediated methylamine utilization pathway in the facultative methylotrophic beta-proteobacteria Methyloversatilis universalis FAM5. Mol. Microbiol. 2010, 75, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.D.; Marx, C.J. Methylamine utilization via the N-methylglutamate pathway in Methylobacterium extorquens PA1 involves a novel flow of carbon through C1 assimilation and dissimilation pathways. J. Bacteriol. 2014, 196, 4130–4139. [Google Scholar] [CrossRef] [PubMed]
- Eady, R.R.; Large, P.J. Purification and properties of methylamine dehydrogenase from Pseudomonas AM1 and its role on growth on methylamine. Biochem. J. 1968, 106, 245–255. [Google Scholar] [PubMed]
- McIntire, W.S.; Wemmer, D.E.; Chistoserdov, A.; Lidstrom, M.E. A new cofactor in a prokaryotic enzyme: Tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase. Science 1991, 252, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Durley, R.; Poliks, B.J.; Hamada, K.; Chen, Z.; Mathews, F.S.; Davidson, V.L.; Satow, Y.; Huizinga, E. Crystal structure of an electron-transfer complex between methylamine dehydrogenase and amicyanin. Biochemistry 1992, 31, 4959–4964. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Davidson, V.L. Purification and properties of methylamine dehydrogenase from Paracoccus denitrificans. J. Bacteriol. 1987, 169, 1712–1717. [Google Scholar] [PubMed]
- Vuilleumier, S.; Chistoserdova, L.; Lee, M.C.; Bringel, F.; Lajus, A.; Zhou, Y.; Gourion, B.; Barbe, V.; Chang, J.; Cruveiller, S.; et al. Methylobacterium genome sequences: A reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. PLoS One 2009, 4, e5584. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Chistoserdova, L.; Lidstrom, M.E. Formaldehyde-detoxification role of the tetrahydromethanopterin-linked pathway in Methylobacterium extorquens AM1. J. Bacteriol. 2003, 197, 7160–7168. [Google Scholar] [CrossRef]
- Chistoserdova, L.; Laukel, M.; Portais, J.C.; Vorholt, J.A.; Lidstrom, M.E. Multiple formate dehydrogenase enzymes in the facultative methylotroph Methylobacterium extorquens AM1 are dispensable for growth on methanol. J. Bacteriol. 2004, 186, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Crowther, G.J.; Vorholt, J.A.; Skovran, E.; Portais, J.C.; Lidstrom, M.E. Identification of a fourth formate dehydrogenase in Methylobacterium extorquens AM1 and confirmation of the essential role of formate oxidation in methylotrophy. J. Bacteriol. 2007, 189, 9076–9081. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Laukel, M.; Vorholt, J.A.; Lidstrom, M.E. Purification of the formate-tetrahydrofolate ligase from Methylobacterium extorquens AM1 and demonstration of its requirement for methylotrophic growth. J. Bacteriol. 2003, 185, 7169–7175. [Google Scholar] [CrossRef] [PubMed]
- Large, P.J.; Quayle, J.R. Microbial growth on C1 compounds. 5. Enzyme activities in extracts of Pseudomonas AM1. Biochem. J. 1963, 87, 386–396. [Google Scholar] [PubMed]
- Chistoserdova, L.; Lidstrom, M.E. Genetics of the serine cycle in Methylobacterium extorquens AM1: Identification of sgaA and mtdA and sequences of sgaA, hprA, and mtdA. J. Bacteriol. 1994, 176, 1957–1968. [Google Scholar] [PubMed]
- Peyraud, R.; Kiefer, P.; Christen, P.; Massou, S.; Portais, J.C.; Vorholt, J.A. Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. PNAS 2009, 106, 4846–4851. [Google Scholar] [CrossRef] [PubMed]
- Gruffaz, C.; Muller, E.E.L.; Louhichi-Jelail, Y.; Nelli, Y.R.; Guichard, G.; Bringel, F. Genes of the N-methylglutamate pathway are essential for growth of Methylobacterium extorquens DM4 with monomethylamine. Appl. Environ. Microbiol. 2014, 80, 3541–3550. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Scanlan, J.; Song, L.; Crombie, A.; Rahman, M.H.; Schäfer, H.; Murrell, J.C. γ-glutamylmethylamide is an essential intermediate in the metabolism of methylamine by Methylocella silvestris. Appl. Environ. Microbiol. 2010, 76, 4530–4537. [Google Scholar] [CrossRef]
- Bellion, E.; Hersh, L.B. Methylamine metabolism in a pseudomonas species. Arch. Biochem. Biophys. 1972, 153, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Pollock, R.J.; Hersh, L.B. N-methylglutamate synthetase: The use of flavin mononucleotide in oxidative catalysis. J. Biol. Chem. 1973, 248, 6724–6733. [Google Scholar] [PubMed]
- Bamforth, C.W.; Large, P.J. Solubilization, partial purification and properties of N-methylglutamate dehydrogenase from Pseudomonas aminovorans. Biochem. J. 1977, 161, 357–370. [Google Scholar] [PubMed]
- Bamforth, C.W.; O’Connor, M.L. The isolation of pleiotropic mutants of Pseudomonas aminovorans deficient in the ability to grow on methylamine and an examination of their enzymic constitution. Microbiology 1979, 110, 143–149. [Google Scholar]
- Chistoserdova, L. Methylotrophy in a lake: From metagenomics to single-organism physiology. Appl. Environ. Microbiol. 2011, 77, 4705–4711. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. Comparative genomics of methylated amine utilization by marine Roseobacter clade bacteria and development if functional gene markers (tmm, gmaS). Environ. Microbiol. 2012, 14, 2308–2322. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Bringel, F.; Chistoserdova, L.; Moulin, L.; Ul Haque, M.F.; Fleischman, D.E.; Gruffaz, C.; Jourand, P.; Knief, C.; Lee, M.C.; et al. Complete genome sequences of six strains of the genus Methylobacterium. J. Bacteriol. 2012, 194, 4746–4748. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.D.; Marx, C.J. Genetic and phenotypic comparison of methylotrophy between Methylobacterium extorquens strains PA1 and AM1. PLoS One 2014, 9, e107887. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.H.; Chiu, H.C.; Delaney, N.F.; Segre, D.; Marx, C.J. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 2011, 332, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Michener, J.K.; Neves, A.A.C.; Vuilleumier, S.; Bringel, F.; Marx, C.J. Effective use of a horizontally-transferred pathway for dichloromethane catabolism requires post-transfer refinement. eLife 2014, 3, 04279. [Google Scholar] [CrossRef]
- Fisher, R.A. The Genetical Theory of Natural Selection; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Kryazhimskiy, S.; Rice, D.P.; Jerison, E.R.; Desai, M.M. Global epistasis makes adaptation predictable despite sequence-level stochasticity. Science 2014, 344, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Agashe, D.; Martinez-Gomez, N.C.; Drummond, D.A.; Marx, C.J. Good codons, bad transcript: Large reductions in gene expression and fitness arising from synonymous mutations in a key enzyme. Mol. Biol. Evol. 2013, 30, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T.A. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, M.O.; Schwartz, R.M. A model of evolutionary change in proteins. Atlas Protein Seq. Struct. 1978, 5, 345–352. [Google Scholar]
- Delaney, N.F.; Kaczmarek, M.E.; Ward, L.M.; Swanson, P.K.; Lee, M.C.; Marx, C.J. Development of an optimized medium, strain, and high-throughput culturing methods for Methylobacterium extorquens. PLoS One 2012, 8, e62957. [Google Scholar] [CrossRef]
- Michener, J.K.; Vuilleumier, S.; Bringel, F.; Marx, C.J. Phylogeny poorly predicts the utility of a challenging horizontally transferred gene in Methylobacterium strains. J. Bacteriol. 2014, 196, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.H.; Marx, C.J. Optimization of gene expression through divergent mutational paths. Cell Rep. 2012, 1, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Chou, H.H.; Marx, C.J. Asymmetric, bimodal trade-offs during adaptation of Methylobacterium to different growth substrates. Evolution 2009, 63, 2816–2830. [Google Scholar] [CrossRef] [PubMed]
- McDonald, I.R.; Doronina, N.V.; Trotsenko, Y.A.; McAnulla, C.; Murrell, J.C. Hyphomicrobium chloromethanicum sp. Nov. and Methylobacterium chloromethanicum sp. Nov., chloromethane-utilizing bacteria isolated from a polluted environment. IJSEM 2001, 51, 119–122. [Google Scholar] [PubMed]
- Gälli, R.; Leisinger, T. Plasmid analysis and cloning of the dichloromethane-utilization genes of Methylobacterium sp. DM4. Microbiology 1988, 134, 943–952. [Google Scholar] [CrossRef]
- Van Aken, B.; Peres, C.M.; Doty, S.L.; Yoon, J.M.; Schnoor, J.L. Methylobacterium populi sp. nov., a novel aerobic, pink-pigmented, facultative methylotrophic, methane-utilizing bacterium isolated from poplar trees (Populus deltoidesxnigra DN34). IJSEM 2004, 54, 1191–1196. [Google Scholar] [PubMed]
- Peel, D.; Quayle, J.R. Microbial growth on C1 compounds: 1. Isolation and characterization of Pseudomonas AM1. Biochem. J. 1961, 81, 465–469. [Google Scholar] [PubMed]
- Knief, C.; Frances, L.; Vorholt, J.A. Competitiveness of diverse Methylobacterium strains in the phyllosphere of Arabidopsis thaliana and identification of representative models, including M. extorquens PA1. Microb. Ecol. 2010, 60, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Delaney, N.F.; Rojas Echenique, J.I.; Marx, C.J. Clarity: An open-source manager for laboratory automation. J. Lab. Autom. 2013, 18, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, C.H.; Hristova, K.; Hanada, S.; Meng, X.Y.; Hanson, J.R.; Scow, K.M.; Kamagata, Y. Methylibium petroleiphilum gen. nov., sp. nov., a novel methyl tert-butyl ether-degrading methylotroph of the Betaproteobacteria. IJSEM 2006, 56, 983–989. [Google Scholar] [PubMed]
- Blount, Z.D.; Borland, C.Z.; Lenski, R.E. Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. PNAS 2008, 105, 7899–7906. [Google Scholar] [CrossRef] [PubMed]
- Vasi, F.; Travisano, M.; Lenski, R.E. Long-term experimental evolution in Escherichia coli. II. Changes in life-history traits during adaptation to a seasonal environment. Am. Nat. 1994, 144, 432–456. [Google Scholar] [CrossRef]
- Blount, Z.D.; Barrick, J.E.; Davidson, C.J.; Lenski, R.E. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature 2012, 489, 513–518. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayak, D.D.; Marx, C.J. Experimental Horizontal Gene Transfer of Methylamine Dehydrogenase Mimics Prevalent Exchange in Nature and Overcomes the Methylamine Growth Constraints Posed by the Sub-Optimal N-Methylglutamate Pathway. Microorganisms 2015, 3, 60-79. https://doi.org/10.3390/microorganisms3010060
Nayak DD, Marx CJ. Experimental Horizontal Gene Transfer of Methylamine Dehydrogenase Mimics Prevalent Exchange in Nature and Overcomes the Methylamine Growth Constraints Posed by the Sub-Optimal N-Methylglutamate Pathway. Microorganisms. 2015; 3(1):60-79. https://doi.org/10.3390/microorganisms3010060
Chicago/Turabian StyleNayak, Dipti D., and Christopher J. Marx. 2015. "Experimental Horizontal Gene Transfer of Methylamine Dehydrogenase Mimics Prevalent Exchange in Nature and Overcomes the Methylamine Growth Constraints Posed by the Sub-Optimal N-Methylglutamate Pathway" Microorganisms 3, no. 1: 60-79. https://doi.org/10.3390/microorganisms3010060
APA StyleNayak, D. D., & Marx, C. J. (2015). Experimental Horizontal Gene Transfer of Methylamine Dehydrogenase Mimics Prevalent Exchange in Nature and Overcomes the Methylamine Growth Constraints Posed by the Sub-Optimal N-Methylglutamate Pathway. Microorganisms, 3(1), 60-79. https://doi.org/10.3390/microorganisms3010060