
Impact of the Gut Microbiota–Metabolite Axis on Intestinal Fatty Acid Absorption in Huainan Pigs

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Sample Collection
2.3. Histological Analysis
2.4. 16S rRNA Sequencing and Analysis
2.5. Untargeted Metabolomics Analysis
2.6. Bioinformatics Analysis
3. Results
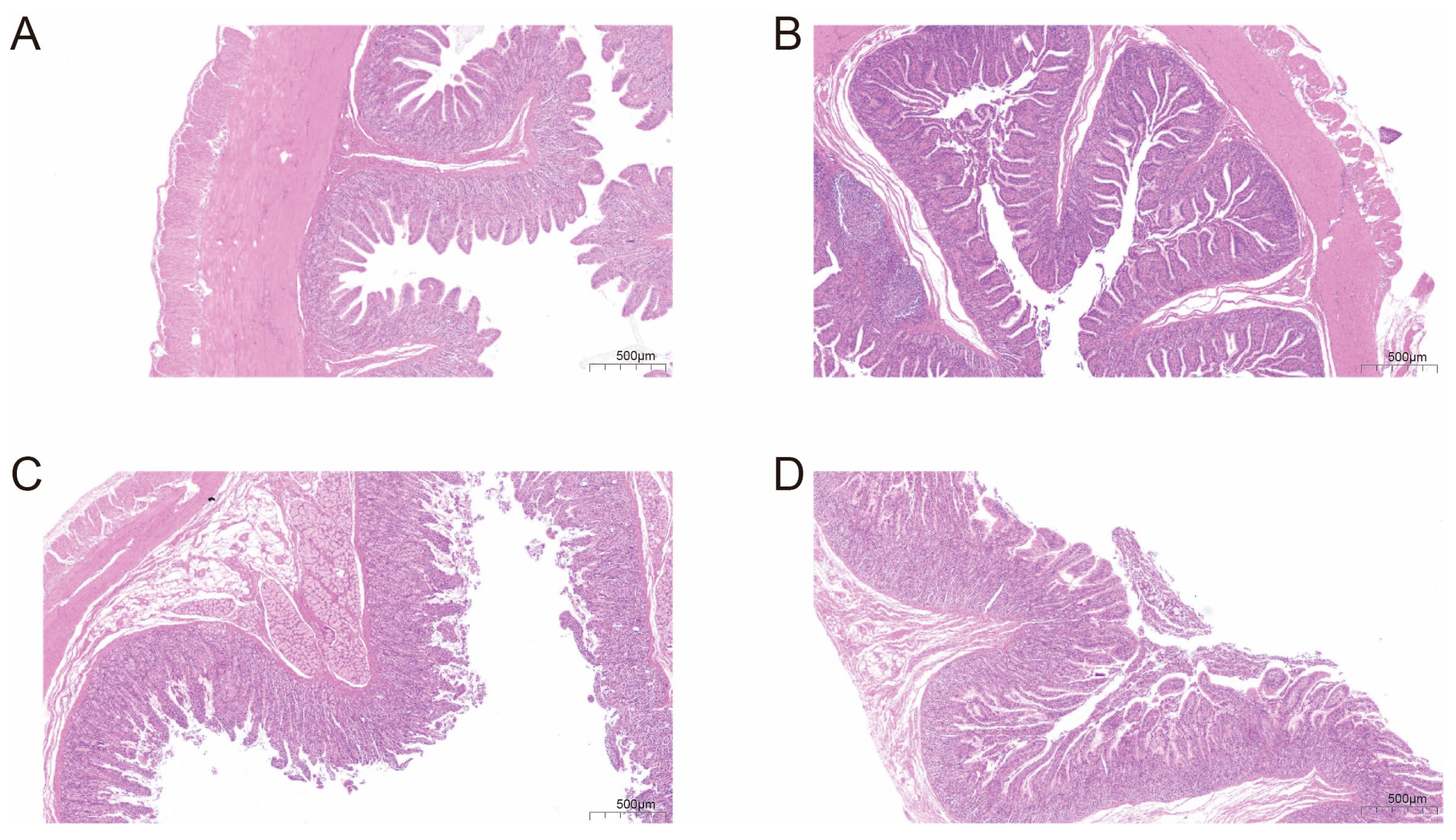
3.1. Intestinal Histological Structure Differs Between Breeds
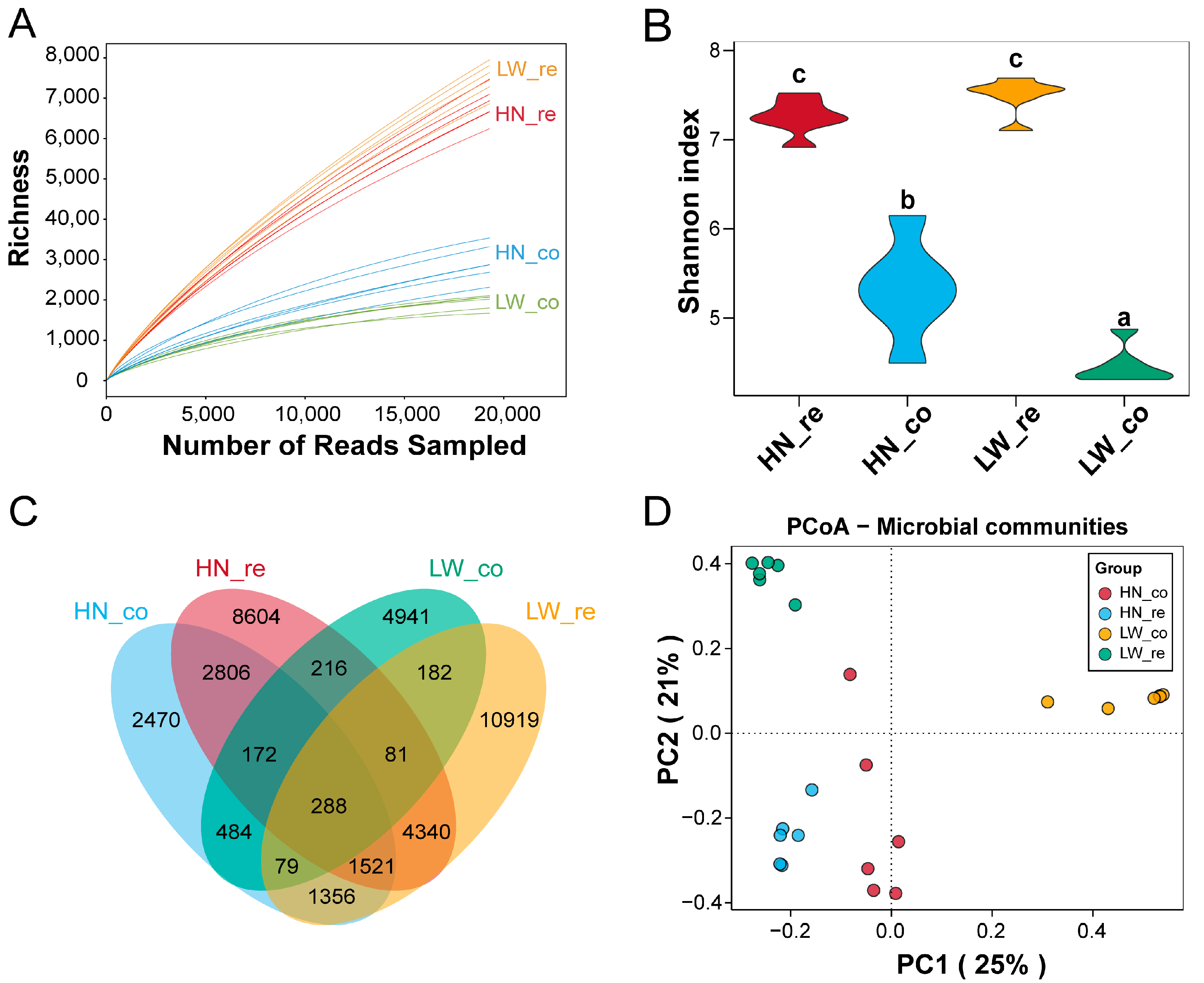
3.2. Segment- and Breed-Specific Microbial Diversity Patterns in the Pig Gut
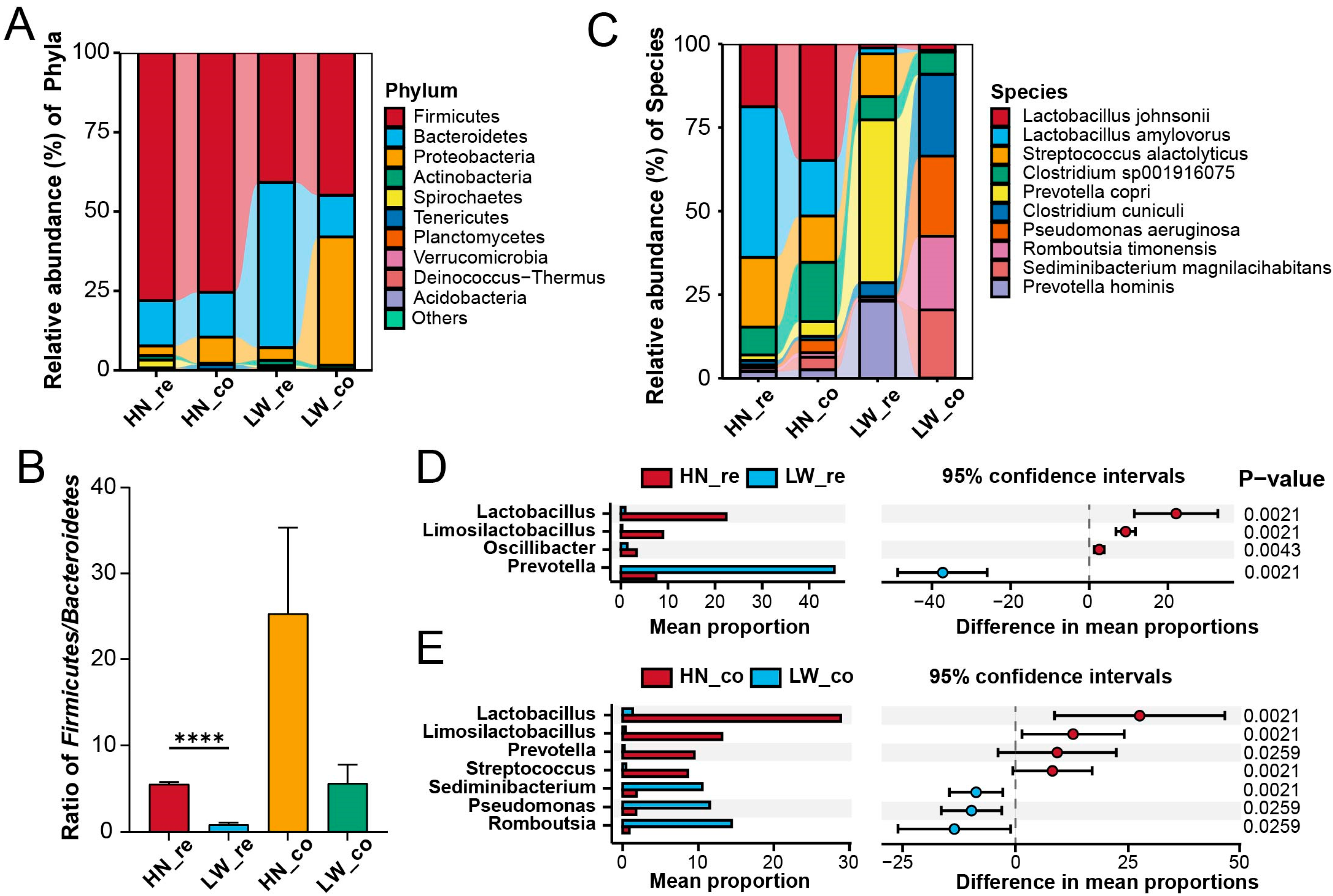
3.3. Compositional Differences in Gut Microbiota Reflect Breed-Specific Metabolic Priorities
3.4. Functional Prediction Reveals Energy-Oriented and Segment-Specific Microbial Capacities in HN Pigs
3.5. Metabolomic Profiling Reveals Breed-Specific Signatures Linked to Lipid Absorption and Immune Modulation
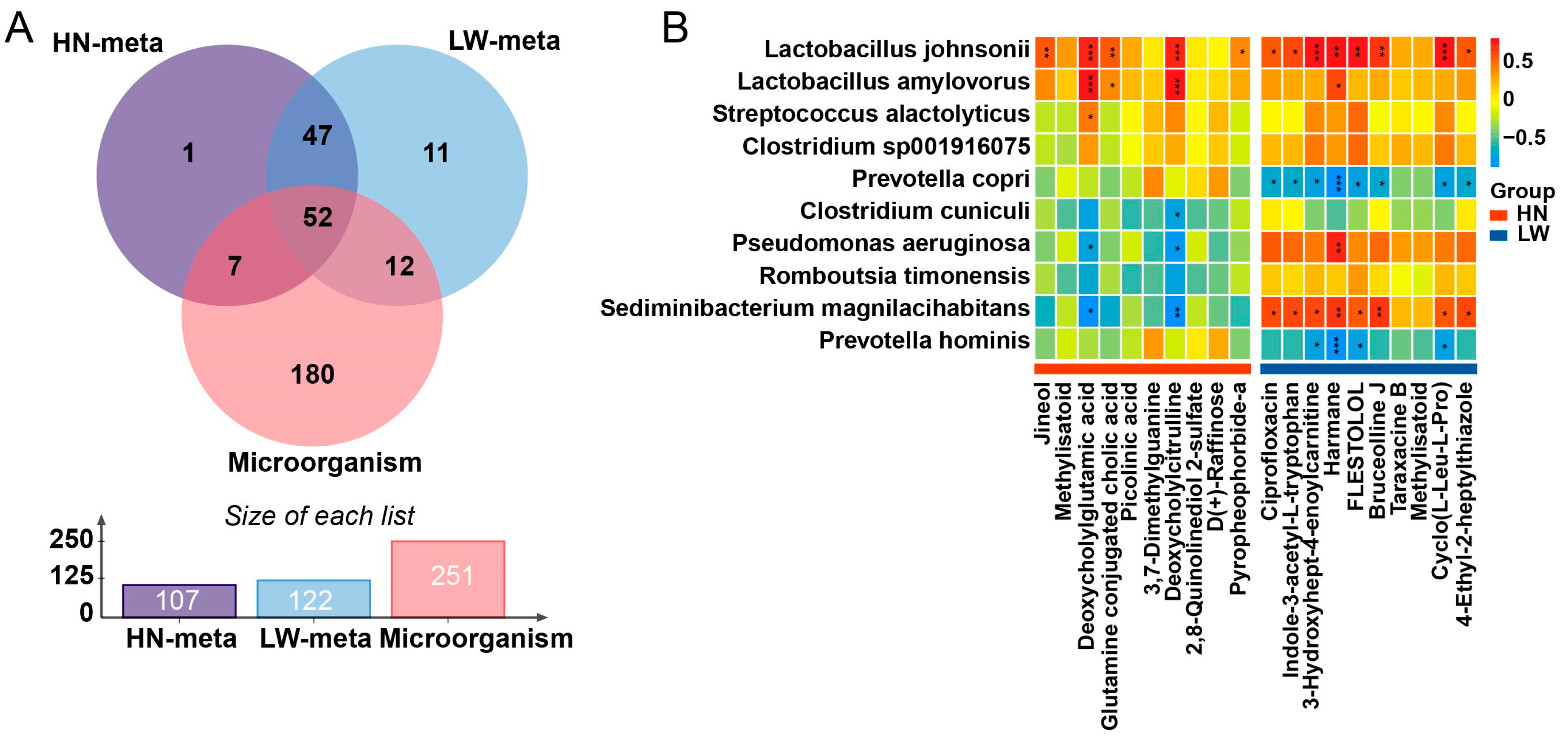
3.6. Integrated Microbiota–Metabolite Analysis Identifies a Coordinated Axis Supporting Lipid Absorption
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC transporter | ATP-Binding Cassette Transporter |
| AUC | Area Under the Curve |
| HN | Huainan pig |
| HMDB | Human Metabolome Database |
| IMF | Intramuscular Fat |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LDA | Linear Discriminant Analysis |
| LEfSe | Linear Discriminant Analysis Effect Size |
| LPS | Lipopolysaccharide |
| LW | Large White pig |
| OPLS-DA | Orthogonal Partial Least Squares Discriminant Analysis |
| OTU | Operational Taxonomic Unit |
| PCoA | Principal Coordinates Analysis |
| PICRUSt2 | Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 |
| PPARγ | Peroxisome Proliferator-Activated Receptor Gamma |
| ROC | Receiver Operating Characteristic |
| SCFA | Short-Chain Fatty Acid |
| VIP | Variable Importance in Projection |
References
- McCormack, U.M.; Curião, T.; Buzoianu, S.G.; Prieto, M.L.; Ryan, T.; Varley, P.; Crispie, F.; Magowan, E.; Metzler-Zebeli, B.U.; Berry, D.; et al. Exploring a Possible Link Between the Intestinal Microbiota and Feed Efficiency in Pigs. Appl. Environ. Microbiol. 2017, 83, e00380-17. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Berri, M.; Estellé, J.; Levenez, F.; Lemonnier, G.; Denis, C.; Leplat, J.J.; Chevaleyre, C.; Billon, Y.; Doré, J.; et al. Early-life establishment of the swine gut microbiome and impact on host phenotypes. Environ. Microbiol. Rep. 2015, 7, 554–569. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.; Kim, E.S.; Keum, G.B.; Doo, H.; Kwak, J.; Pandey, S.; Cho, J.H.; Ryu, S.; Song, M.; et al. Comparative analysis of the pig gut microbiome associated with the pig growth performance. J. Anim. Sci. Technol. 2023, 65, 856–864. [Google Scholar] [CrossRef]
- Zhang, Q.; Du, M.; Wei, S.; Zhu, L.; Yan, R.; Jin, M.; Wang, Y. Variation of meat quality and relationship to gut microbiota among different pig breeds. Microb. Biotechnol. 2025, 18, e70139. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, M.; Chen, J.; Li, Y.; Kuang, Z.; Dende, C.; Raj, P.; Quinn, G.; Hu, Z.; Srinivasan, T.; et al. The gut microbiota reprograms intestinal lipid metabolism through long noncoding RNA Snhg9. Science 2023, 381, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, G.E.; Metzler-Zebeli, B.U.; Lawlor, P.G. Impact of intestinal microbiota on growth and feed efficiency in pigs: A review. Microorganisms 2020, 8, 1886. [Google Scholar] [CrossRef]
- Ma, J.; Duan, Y.; Li, R.; Liang, X.; Li, T.; Huang, X.; Yin, Y.; Yin, J. Gut microbial profiles and the role in lipid metabolism in Shaziling pigs. Anim. Nutr. 2022, 9, 345–356. [Google Scholar] [CrossRef]
- Bergamaschi, M.; Tiezzi, F.; Howard, J.; Huang, Y.J.; Gray, K.A.; Schillebeeckx, C.; McNulty, N.P.; Maltecca, C. Gut microbiome composition differences among breeds impact feed efficiency in swine. Microbiome 2020, 8, 110. [Google Scholar] [CrossRef]
- Chen, H.; Nwe, P.K.; Yang, Y.; Rosen, C.E.; Bielecka, A.A.; Kuchroo, M.; Cline, G.W.; Kruse, A.C.; Ring, A.M.; Crawford, J.M.; et al. A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell 2019, 177, 1217–1231. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, L.; Liu, Z.; Guo, A.; Yang, G.; Yu, T. Host-microbiota interactions in the pathogenesis of porcine fetal mummification. Microorganisms 2025, 13, 1052. [Google Scholar] [CrossRef]
- Colosimo, D.A.; Kohn, J.A.; Luo, P.M.; Piscotta, F.J.; Han, S.M.; Pickard, A.J.; Rao, A.; Cross, J.R.; Cohen, L.J.; Brady, S.F.; et al. Mapping interactions of microbial metabolites with human G-protein-coupled receptors. Cell Host Microbe 2019, 26, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Zhao, S.; Su, Z.; Ge, C.; Zhang, Y.; Jia, J.; et al. The intestinal microbiome associated with lipid metabolism and obesity in humans and animals. J. Appl. Microbiol. 2022, 133, 2915–2930. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.Y.P.; Visvalingam, V.; Wahli, W. The PPAR-microbiota-metabolic organ trilogy to fine-tune physiology. FASEB J. 2019, 33, 9706–9730. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, S.H.; Li, H.L.; Zhou, X.B.; Zhou, L.W.; Chen, C.; Mansell, T.; Novakovic, B.; Saffery, R.; Baker, P.N.; et al. The attenuation of gut microbiota-derived short-chain fatty acids elevates lipid transportation through suppression of the intestinal HDAC3-H3K27ac-PPAR-γ axis in gestational diabetes mellitus. J. Nutr. Biochem. 2024, 133, 109708. [Google Scholar] [CrossRef]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef]
- Du, J.; Gan, M.; Xie, Z.; Du, G.; Luo, Y.; Liu, B.; Zhu, K.; Chen, L.; Zhao, Y.; Niu, L.; et al. A comparative study on the growth performance and gut microbial composition of Duroc and Yorkshire boars. Genes 2023, 14, 1726. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Cheng, M.; Ye, F.; Li, W.; Wang, C.; Huang, Y.; Wu, Y.; Xuan, R.; Liu, G.; et al. Gut microbial diversity among Yorkshire, Landrace and Duroc boars and its impact on semen quality. AMB Express 2022, 12, 158. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.; Zhang, C.; Wang, G.; Shi, C.; Li, Z.; Gao, F.; Cui, Y.; Li, M.; Yang, G. Composition and evolutionary characterization of the gut microbiota in pigs. Int. Microbiol. 2024, 27, 993–1008. [Google Scholar] [CrossRef]
- Wang, W.; Wang, D.; Zhang, X.; Liu, X.; Niu, X.; Li, S.; Huang, S.; Ran, X.; Wang, J. Comparative transcriptome analysis of longissimus dorsi muscle reveal potential genes affecting meat trait in Chinese indigenous Xiang pig. Sci. Rep. 2024, 14, 8486. [Google Scholar] [CrossRef]
- Murga-Garrido, S.M.; Hong, Q.; Cross, T.L.; Hutchison, E.R.; Han, J.; Thomas, S.P.; Vivas, E.I.; Denu, J.; Ceschin, D.G.; Tang, Z.Z.; et al. Gut microbiome variation modulates the effects of dietary fiber on host metabolism. Microbiome 2021, 9, 117. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, J.; Zhao, Y.; Lin, Z.; Ji, L.; Ma, X. Tibetan pig-derived probiotic Lactobacillus amylovorus SLZX20-1 improved intestinal function via producing enzymes and regulating intestinal microflora. Front. Nutr. 2022, 9, 846991. [Google Scholar] [CrossRef] [PubMed]
- GB/T 42304-2023; Welfare Criteria for Animals to Be Slaughtered. State Administration for Market Regulation, Standardization Administration of the People’s Republic of China: Beijing, China, 2023.
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Wright, R.J.; Langille, M.G.I. PICRUSt2-SC: An update to the reference database used for functional prediction within PICRUSt2. Bioinformatics 2025, 41, btaf269. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Shokryazdan, P.; Sieo, C.C.; Kalavathy, R.; Liang, J.B.; Alitheen, N.B.; Faseleh Jahromi, M.; Ho, Y.W. Probiotic potential of Lactobacillus strains with antimicrobial activity against some human pathogenic strains. Biomed Res. Int. 2014, 2014, 927268. [Google Scholar] [CrossRef]
- Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An aPPARent functional consequence in skeletal muscle physiology via peroxisome proliferator-activated receptors. Int. J. Mol. Sci. 2018, 19, 1425. [Google Scholar] [CrossRef]
- Kataoka, K. The intestinal microbiota and its role in human health and disease. J. Med. Invest. 2016, 63, 27–37. [Google Scholar] [CrossRef]
- Egamberdiyeva, D. Characterization of Pseudomonas species isolated from the rhizosphere of plants grown in serozem soil, semi arid region of Uzbekistan. Sci. World J. 2005, 5, 501–509. [Google Scholar] [CrossRef]
- Pu, G.; Hou, L.; Du, T.; Zhou, W.; Liu, C.; Niu, P.; Wu, C.; Bao, W.; Huang, R.; Li, P. Increased proportion of fiber-degrading microbes and enhanced cecum development jointly promote host to digest appropriate high-fiber diets. mSystems 2023, 8, e0093722. [Google Scholar] [CrossRef]
- Lacourt, T.E.; Vichaya, E.G.; Chiu, G.S.; Dantzer, R.; Heijnen, C.J. The high costs of low-grade inflammation: Persistent fatigue as a consequence of reduced cellular-energy availability and non-adaptive energy expenditure. Front. Behav. Neurosci. 2018, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cano, M.M.; González-Gómez, M.J.; Sánchez-Solana, B.; Monsalve, E.M.; Díaz-Guerra, M.M.; Laborda, J.; Nueda, M.L.; Baladrón, V. NOTCH receptors and DLK proteins enhance brown adipogenesis in mesenchymal C3H10T1/2 cells. Cells 2020, 9, 2032. [Google Scholar] [CrossRef] [PubMed]
- Uno, Y.; Morikuni, S.; Shiraishi, M.; Asano, A.; Kawaguchi, H.; Murayama, N.; Yamazaki, H. A comprehensive analysis of six forms of cytochrome P450 2C (CYP2C) in pigs. Xenobiotica 2022, 52, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Up to date on cholesterol 7 alpha-hydroxylase (CYP7A1) in bile acid synthesis. Liver Res. 2020, 4, 47–63. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y. Gut microbiota-derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef]
- Lyu, M.; Bai, Y.; Orihara, K.; Miyanaga, K.; Yamamoto, N. GAPDH released from Lactobacillus johnsonii MG enhances barrier function by upregulating genes associated with tight junctions. Microorganisms 2023, 11, 1393. [Google Scholar] [CrossRef]
- Jin, S.J.; Song, Y.; Park, H.S.; Park, K.W.; Lee, S.; Kang, H. Harmine inhibits multiple TLR-induced inflammatory expression through modulation of NF-κB p65, JNK, and STAT1. Life 2022, 12, 2022. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Zhu, L.; Wang, Y.; Ma, Q.; Yan, X.; Li, M.; Xing, B. Impact of the Gut Microbiota–Metabolite Axis on Intestinal Fatty Acid Absorption in Huainan Pigs. Microorganisms 2025, 13, 1609. https://doi.org/10.3390/microorganisms13071609
Wang J, Zhu L, Wang Y, Ma Q, Yan X, Li M, Xing B. Impact of the Gut Microbiota–Metabolite Axis on Intestinal Fatty Acid Absorption in Huainan Pigs. Microorganisms. 2025; 13(7):1609. https://doi.org/10.3390/microorganisms13071609
Chicago/Turabian StyleWang, Jing, Liangying Zhu, Yangyang Wang, Qiang Ma, Xiangzhou Yan, Mingxun Li, and Baosong Xing. 2025. "Impact of the Gut Microbiota–Metabolite Axis on Intestinal Fatty Acid Absorption in Huainan Pigs" Microorganisms 13, no. 7: 1609. https://doi.org/10.3390/microorganisms13071609
APA StyleWang, J., Zhu, L., Wang, Y., Ma, Q., Yan, X., Li, M., & Xing, B. (2025). Impact of the Gut Microbiota–Metabolite Axis on Intestinal Fatty Acid Absorption in Huainan Pigs. Microorganisms, 13(7), 1609. https://doi.org/10.3390/microorganisms13071609