

Phylogroup Homeostasis of Escherichia coli in the Human Gut Reflects the Physiological State of the Host


Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets Used for Intraspecific Taxonomic Analysis
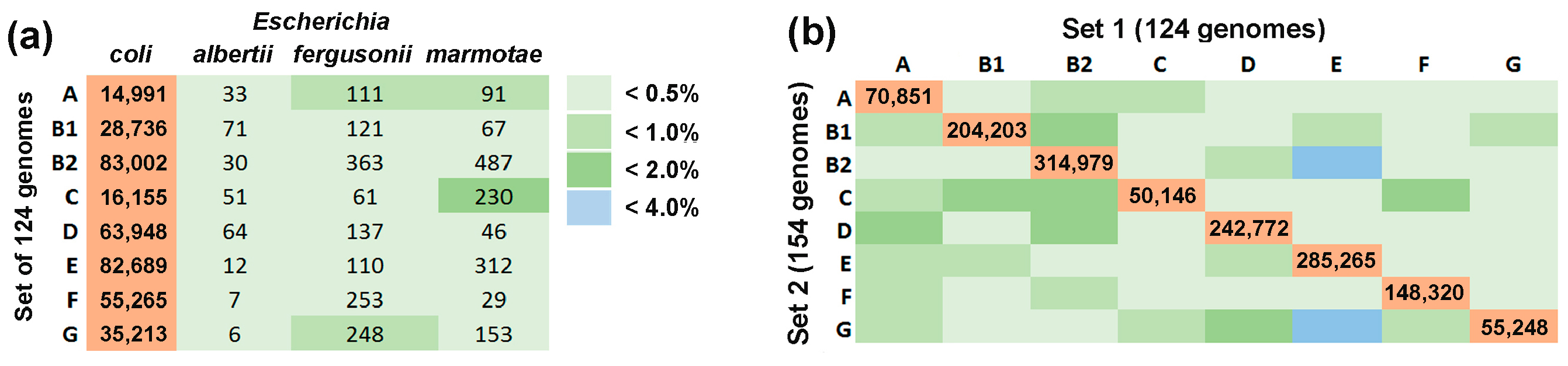
2.2. Genome Collections
2.3. Barcoding of the E. coli Phylogroups
2.4. Alternative Intraspecific Phylotyping of E. coli
2.5. Analysis of Human Metagenomes Based on Phylogroup-Specific Taxonomy
2.6. Machine Learning Methods for Binary Clustering
2.7. Dataset Visualization
2.8. Statistical Analysis
3. Results
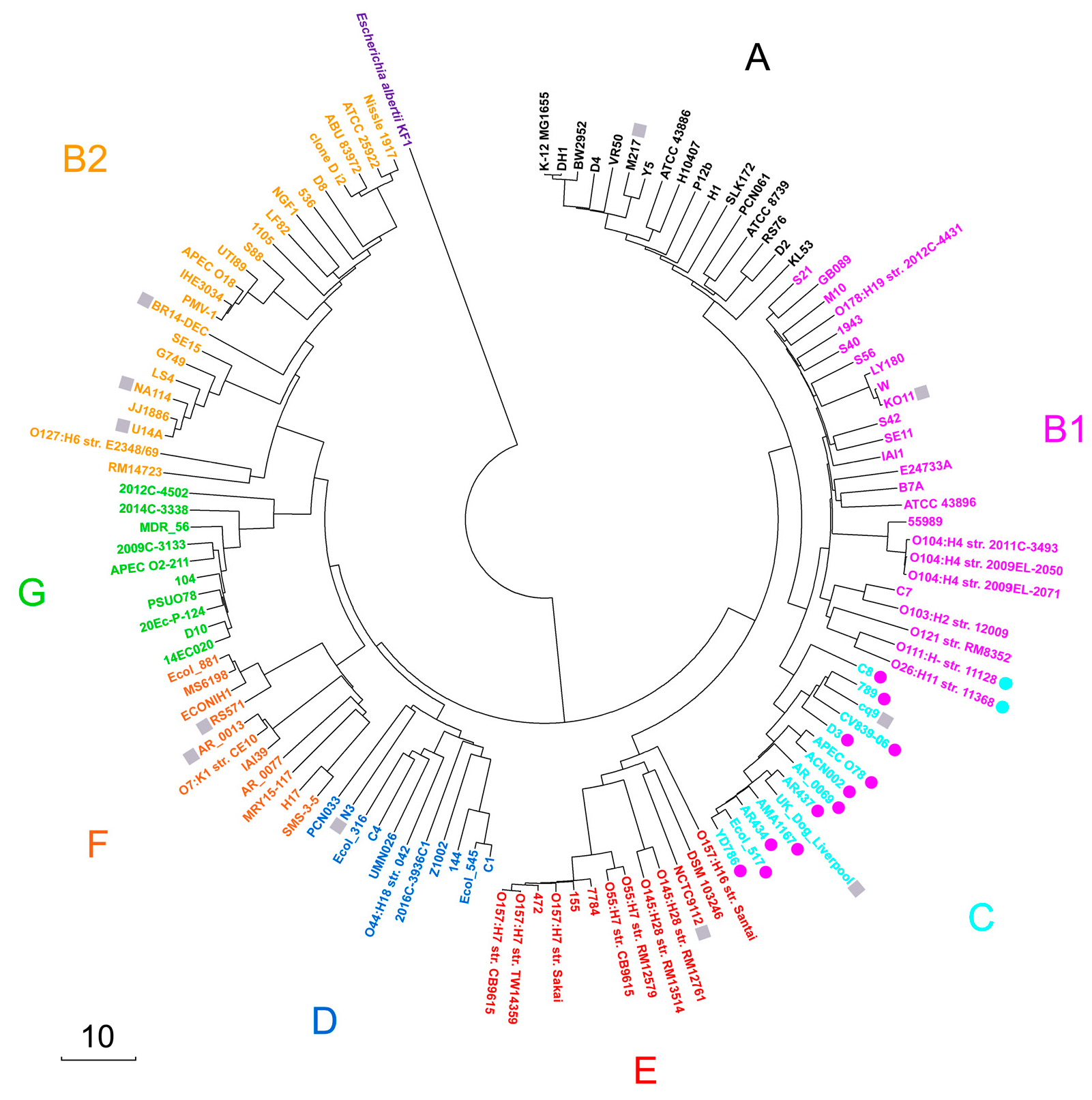
3.1. Validation of Intraspecific E. coli Phylotyping Using MASH-like Option of UniSeq Pipeline
3.2. Assessment of Barcoding Specificity
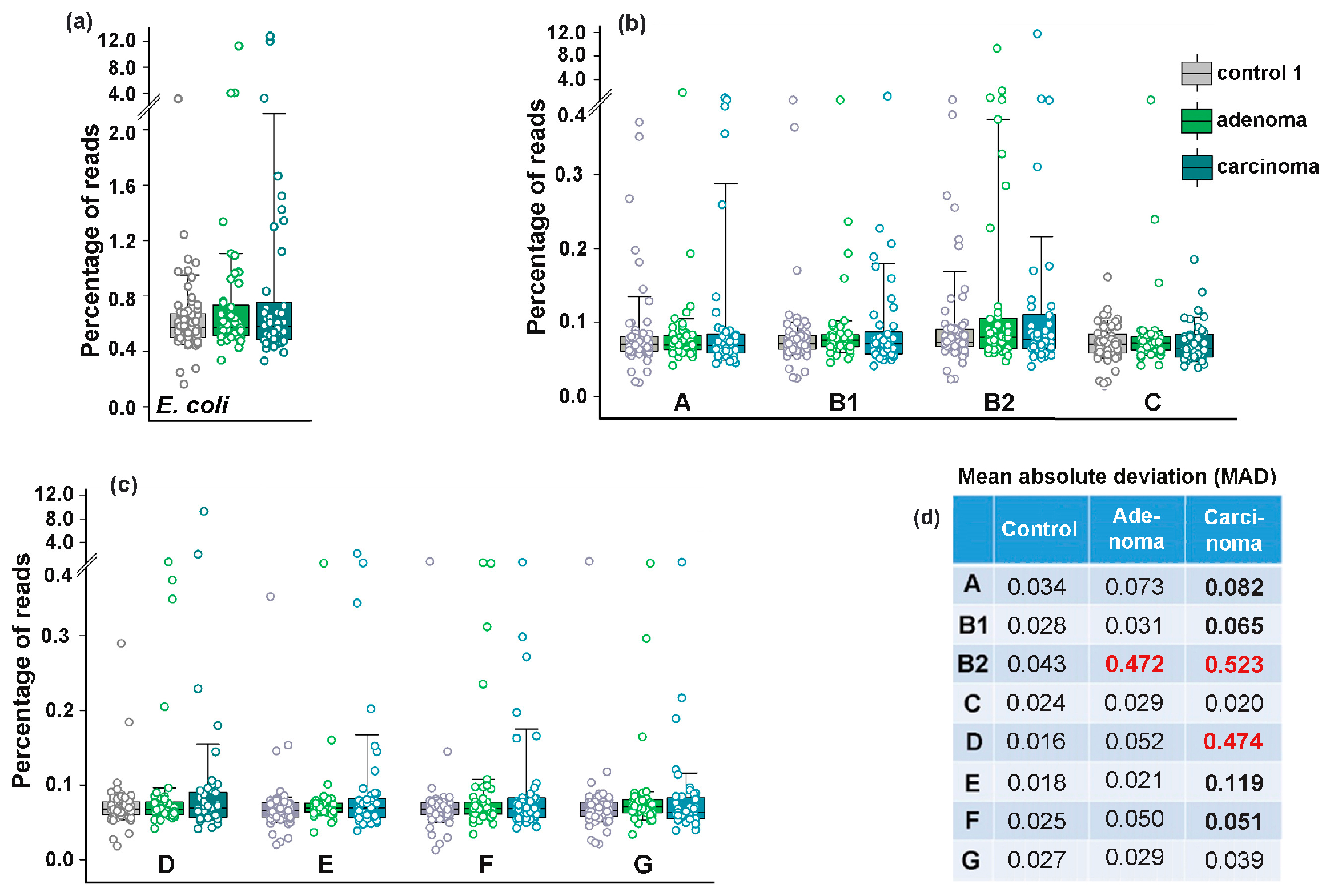
3.3. Colorectal Adenoma and Carcinoma Had Different Impact on the Distribution of E. coli Phylogroups in the Intestinal Microflora
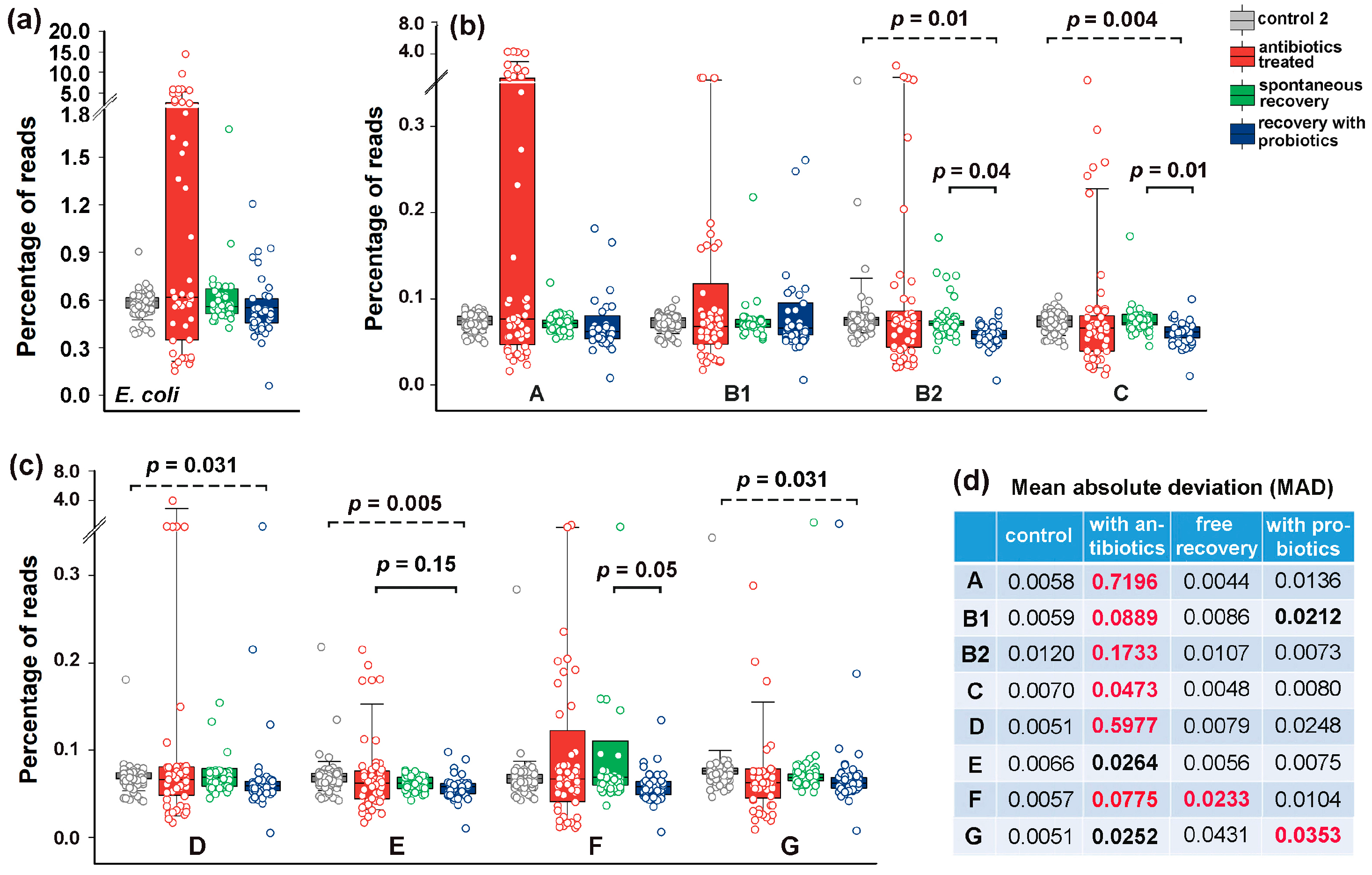
3.4. Bimodal Response of E. coli to Antibiotics and Recovery with Probiotics
3.5. The Response of E. coli to Antibiotics Was Not Uniform Among Phylogroups
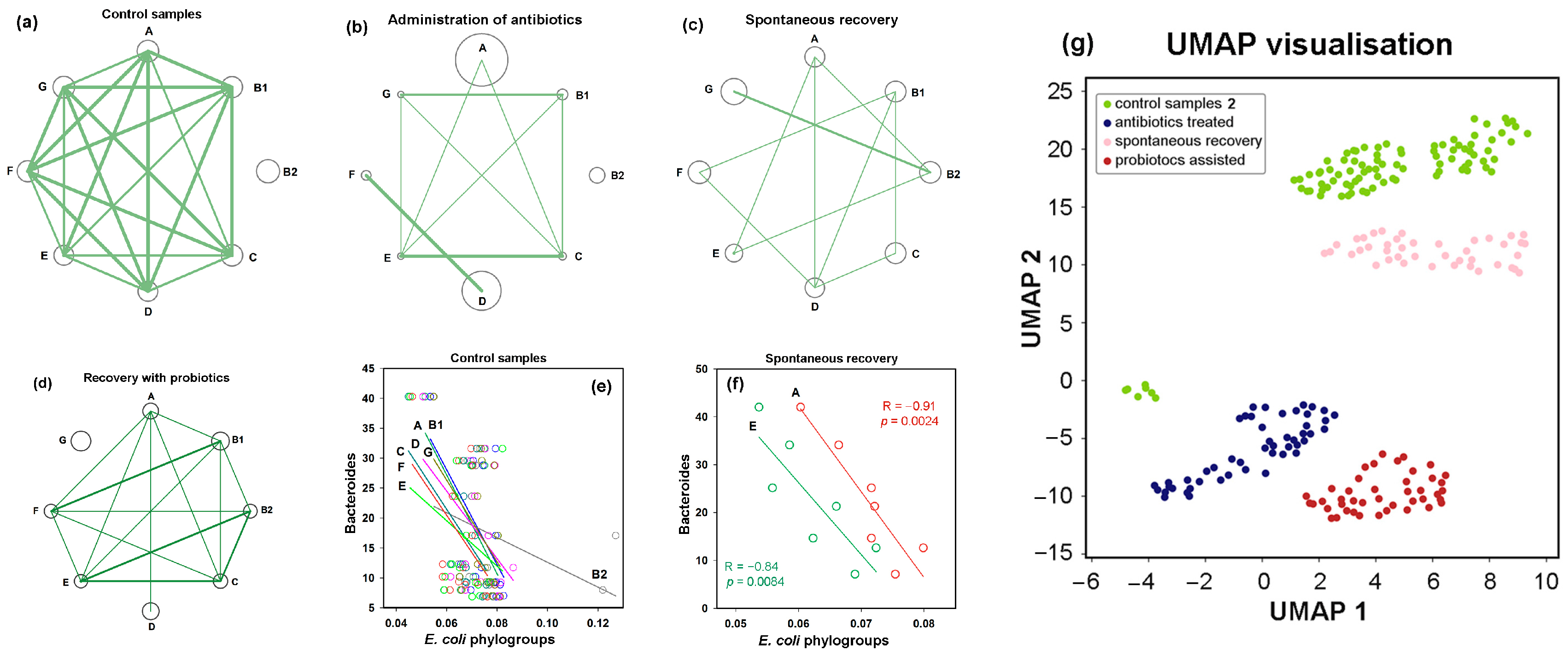
3.6. Post-Treatment Recovery Partially Restored the Intra- and Interspecies Balance Disrupted by Antibiotics, but Not the Original Correlation Between Phylogroups and the UMAP Cluster
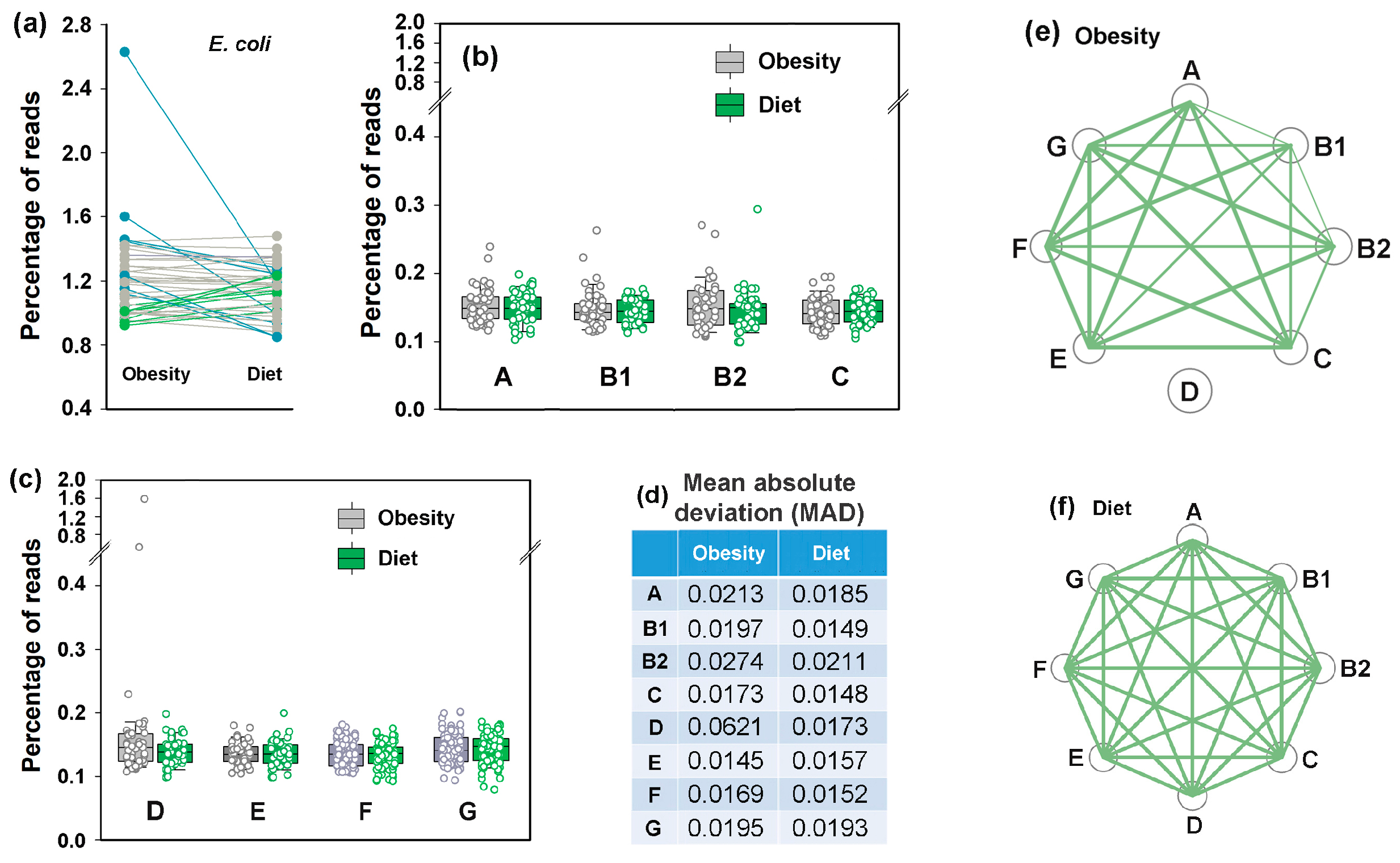
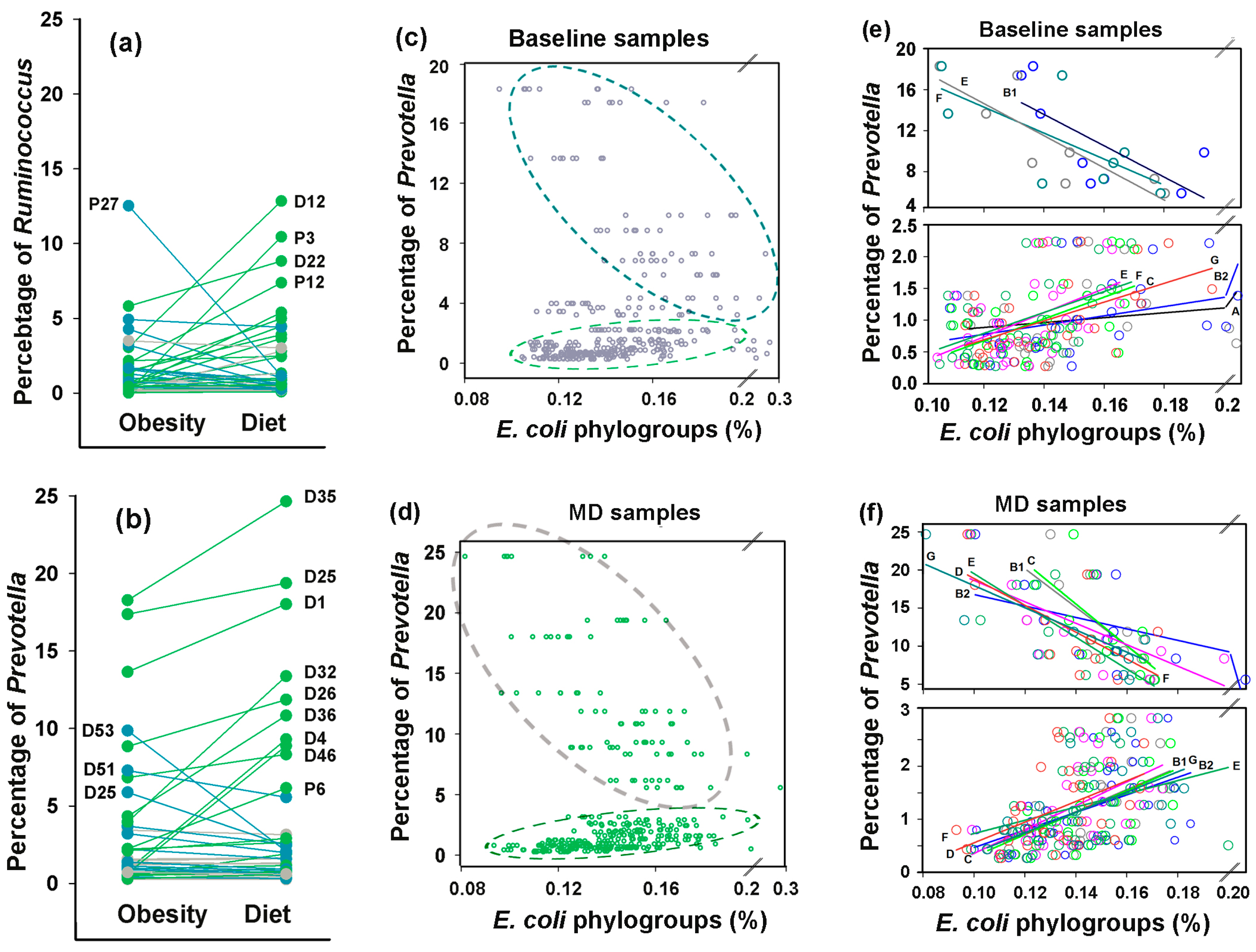
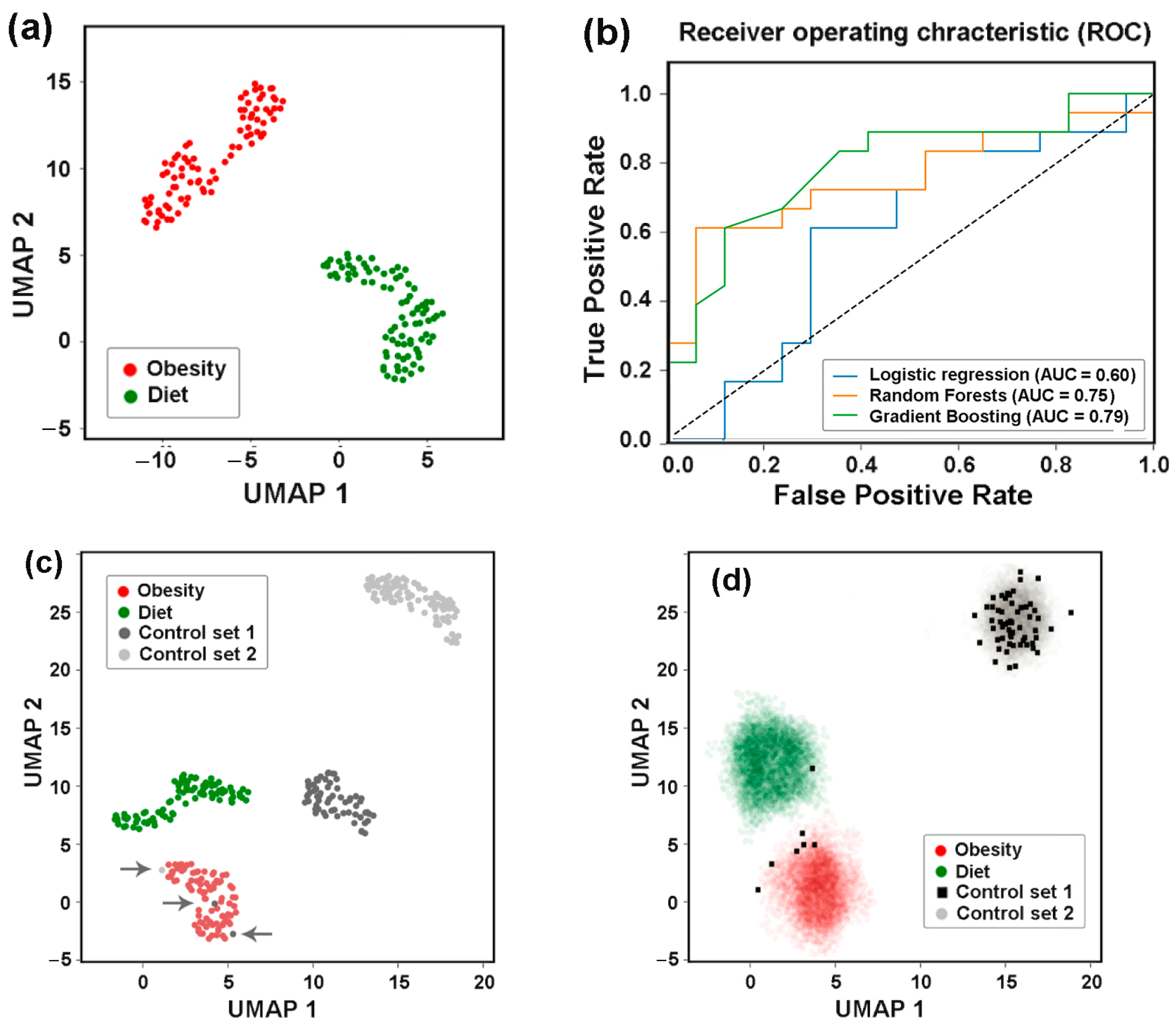
3.7. Idealizing E. coli Intraspecific Balance, the Mediterranean Diet Intensified Its Negative Link to Bacteroides and Unlocked Bidirectional Connections with Prevotella
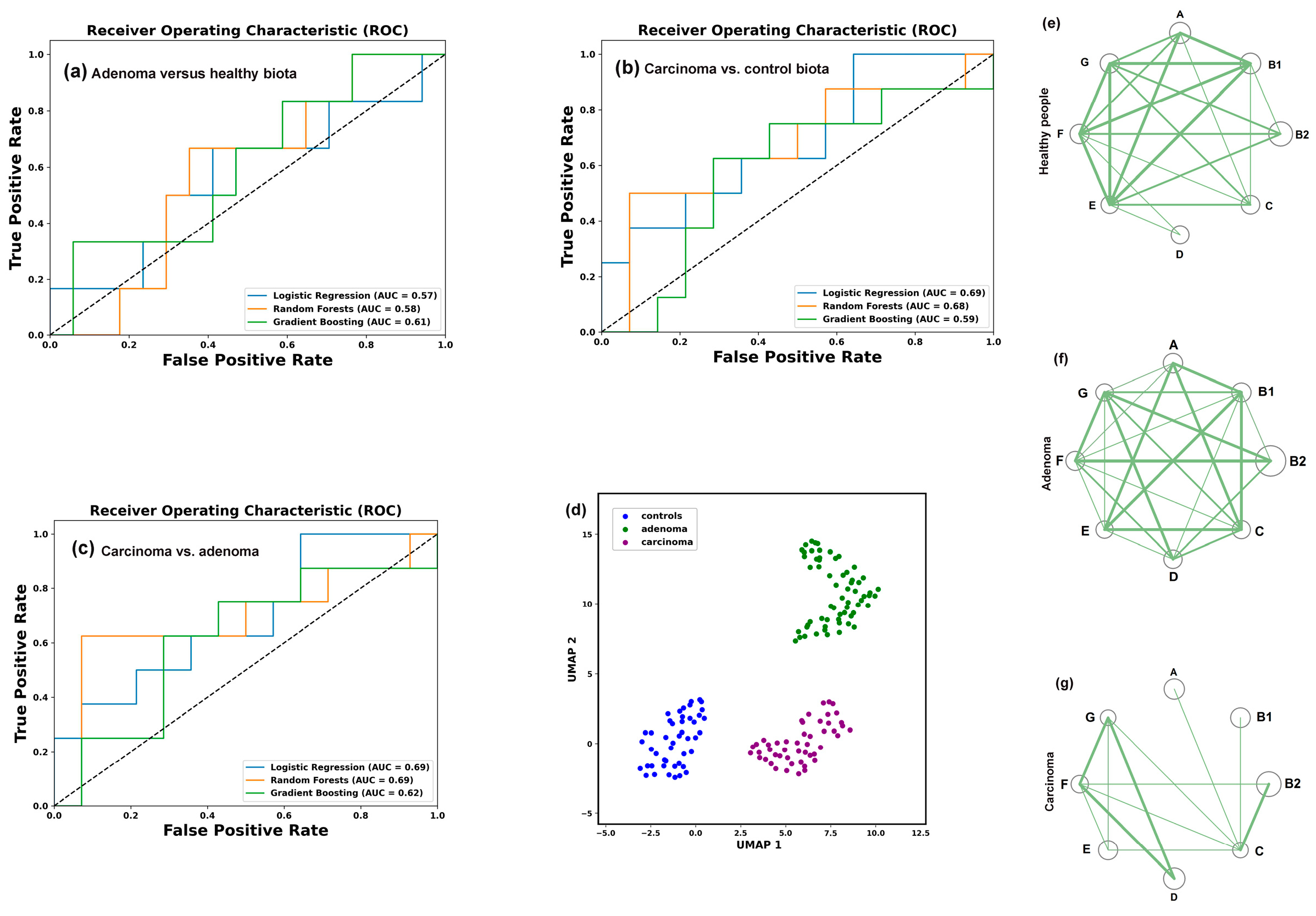
3.8. Assessing Difference Between Samples, Machine Learning Approaches May Also Be Implemented to Reveal Individual Similarity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Martinson, J.N.V.; Walk, S.T. Escherichia coli residency in the gut of healthy human adults. EcoSal Plus 2020, 9, ESP-0003-2020. [Google Scholar] [CrossRef]
- Clermont, O.; Bonacorsi, S.; Bingen, E. Rapid and simple determination of the Escherichia coli phylogenetic group. Appl. Environ. Microbiol. 2000, 66, 4555–4558. [Google Scholar] [CrossRef]
- Jaureguy, F.; Landraud, L.; Passet, V.; Diancourt, L.; Frapy, E.; Guigon, G.; Carbonnelle, E.; Lortholary, O.; Clermont, O.; Denamur, E.; et al. Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains. BMC Genom. 2008, 9, 560. [Google Scholar] [CrossRef]
- Clermont, O.; Olier, M.; Hoede, C.; Diancourt, L.; Brisse, S.; Keroudean, M.; Glodt, J.; Picard, B.; Oswald, E.; Denamur, E. Animal and human pathogenic Escherichia coli strains share common genetic backgrounds. Infect. Genet. Evol. 2011, 11, 654–662. [Google Scholar] [CrossRef]
- Beghain, J.; Bridier-Nahmias, A.; Le Nagard, H.; Denamur, E.; Clermont, O. ClermonTyping: An easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 2018, 4, e000192. [Google Scholar] [CrossRef]
- Clermont, O.; Christenson, J.K.; Denamur, E.; Gordon, D.M. The Clermont Escherichia coli phylo-typing method revisited: Improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 2013, 5, 58–65. [Google Scholar] [CrossRef]
- Lu, S.; Jin, D.; Wu, S.; Yang, J.; Lan, R.; Bai, X.; Liu, S.; Meng, Q.; Yuan, X.; Zhou, J.; et al. Insights into the evolution of pathogenicity of Escherichia coli from genomic analysis of intestinal E. coli of Marmota himalayana in Qinghai-Tibet plateau of China. Emerg. Microbes Infect. 2016, 5, e122. [Google Scholar] [CrossRef]
- Clermont, O.; Dixit, O.V.A.; Vangchhia, B.; Condamine, B.; Dion, S.; Bridier-Nahmias, A.; Denamur, E.; Gordon, D. Characterization and rapid identification of phylogroup G in Escherichia coli, a lineage with high virulence and antibiotic resistance potential. Environ. Microbiol. 2019, 21, 3107–3117. [Google Scholar] [CrossRef]
- Panyukov, V.V.; Kiselev, S.S.; Ozoline, O.N. Unique k-mers as strain-specific barcodes for phylogenetic analysis and natural microbiome profiling. Int. J. Mol. Sci. 2020, 21, 944. [Google Scholar] [CrossRef]
- Abram, K.; Udaondo, Z.; Bleker, C.; Wanchai, V.; Wassenaar, T.M.; Robeson, M.S.; Ussery, D.W. Mash-based analyses of Escherichia coli genomes reveal 14 distinct phylogroups. Commun. Biol. 2021, 4, 117. [Google Scholar] [CrossRef]
- Picard, B.; Garcia, J.S.; Gouriou, S.; Duriez, P.; Brahimi, N.; Bingen, E.; Elion, J.; Denamur, E. The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect. Immun. 1999, 67, 546–553. [Google Scholar] [CrossRef]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef]
- Escobar-Paramo, P.; Clermont, O.; Blanc-Potard, A.B.; Bui, H.; Le Bouguenec, C.; Denamur, E. A specific genetic background is required for acquisition and expression of virulence factors in Escherichia coli. Mol. Biol. Evol. 2004, 21, 1085–1094. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Chakraborty, A.; Saralaya, V.; Adhikari, P.; Shenoy, S.; Baliga, S.; Hegde, A. Characterization of Escherichia coli phylogenetic groups associated with extraintestinal infections in South Indian population. Ann. Med. Health Sci. Res. 2015, 5, 241–246. [Google Scholar] [CrossRef]
- Rasko, D.A.; Webster, D.R.; Sahl, J.W.; Bashir, A.; Boisen, N.; Scheutz, F.; Paxinos, E.E.; Sebra, R.; Chin, C.-S.; Iliopoulos, D.; et al. Origins of the E. coli strain causing an outbreak of hemolytic–uremic syndrome in Germany. N. Engl. J. Med. 2011, 365, 709–717. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Mellmann, A.; Zhang, W.; Kock, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: A microbiological study. Lancet Infect. Dis. 2011, 11, 671–676. [Google Scholar] [CrossRef]
- Brzuszkiewicz, E.; Thurmer, A.; Schuldes, J.; Leimbach, A.; Liesegang, H.; Meyer, F.D.; Boelter, J.; Petersen, H.; Gottschalk, G.; Daniel, R. Genome sequence analyses of two isolates from the recent Escherichia coli outbreak in Germany reveal the emergence of a new pathotype: Entero-Aggregative-Haemorrhagic Escherichia coli (EAHEC). Arch. Microbiol. 2011, 193, 883–891. [Google Scholar] [CrossRef]
- Schiller, P.; Knodler, M.; Berger, P.; Greune, L.; Fruth, A.; Mellmann, A.; Dersch, P.; Berger, M.; Dobrindt, U. The superior adherence phenotype of E. coli O104:H4 is directly mediated by the aggregative adherence fimbriae type I. Virulence 2021, 12, 346–359. [Google Scholar] [CrossRef]
- Burger, R. EHEC O104:H4 in Germany 2011: Large outbreak of bloody diarrhea and haemolytic uraemic syndrome by shiga toxin–producing E. coli via contaminated food. In Improving Food Safety Through a One Health Approach: Workshop Summary; Choffnes, E.R., Relman, D.A., Olsen, L., Hutton, R., Mack, A., Eds.; National Academies Press: Washington, DC, USA, 2012; pp. 115–129. [Google Scholar]
- Putonti, C.; Luo, Y.; Katili, C.; Chumakov, S.; Fox, G.E.; Graur, D.; Fofanov, Y. A computational tool for the genomic identification of regions of unusual compositional properties and its utilization in the detection of horizontally transferred sequences. Mol. Biol. Evol. 2006, 23, 1863–1868. [Google Scholar] [CrossRef]
- Lim, J.Y.; Yoon, J.; Hovde, C.J. A brief overview of Escherichia coli O157:H7 and its plasmid O157. J. Microbiol. Biotechnol. 2010, 20, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Wick, L.M.; Qi, W.; Lacher, D.W.; Whittam, T.S. Evolution of genomic content in the stepwise emergence of Escherichia coli O157:H7. J. Bacteriol. 2005, 187, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Rahal, E.A.; Kazzi, N.; Nassar, F.J.; Matar, G.M. Escherichia coli O157:H7–clinical aspects and novel treatment approaches. Front. Cell. Infect. Microbiol. 2012, 2, 138. [Google Scholar] [CrossRef] [PubMed]
- Franzin, F.M.; Sircili, M.P. Locus of enterocyte effacement: A pathogenicity island involved in the virulence of enteropathogenic and enterohemorrhagic Escherichia coli subjected to a complex network of gene regulation. Biomed. Res. Int. 2015, 2015, 534738. [Google Scholar] [CrossRef]
- Perna, N.T.; Plunkett, G.; Burland, V.; Mau, B.; Glasner, J.D.; Rose, D.J.; Mayhew, G.F.; Evans, P.S.; Gregor, J.; Kirkpatrick, H.A.; et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 2001, 409, 529–533. [Google Scholar] [CrossRef]
- Hayashi, T.; Makino, K.; Ohnishi, M.; Kurokawa, K.; Ishii, K.; Yokoyama, K.; Han, C.G.; Ohtsubo, E.; Nakayama, K.; Murata, T.; et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001, 8, 11–22. [Google Scholar] [CrossRef]
- Jiang, L.; Yang, W.; Jiang, X.; Yao, T.; Wang, L.; Yang, B. Virulence-related O islands in enterohemorrhagic Escherichia coli O157:H7. Gut Microbes 2021, 13, e1992237. [Google Scholar] [CrossRef]
- Taieb, F.; Petit, C.; Nougayrede, J.P.; Oswald, E. The enterobacterial genotoxins: Cytolethal distending toxin and colibactin. EcoSal Plus 2016, 7, ESP-0008-2016. [Google Scholar] [CrossRef]
- Nougayrede, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- Kruis, W.; Fric, P.; Pokrotnieks, J.; Lukas, M.; Fixa, B.; Kascak, M.; Kamm, M.A.; Weismueller, J.; Beglinger, C.; Stolte, M.; et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 2004, 53, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Dubbert, S.; Klinkert, B.; Schimiczek, M.; Wassenaar, T.M.; Bunau, R.V. No genotoxicity is detectable for Escherichia coli strain Nissle 1917 by standard in vitro and in vivo tests. Eur. J. Microbiol. Immunol. 2020, 10, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Homburg, S.; Oswald, E.; Hacker, J.; Dobrindt, U. Expression analysis of the colibactin gene cluster coding for a novel polyketide in Escherichia coli. FEMS Microbiol. Lett. 2007, 275, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.M. E. coli and colorectal cancer: A complex relationship that deserves a critical mindset. Crit. Rev. Microbiol. 2018, 44, 619–632. [Google Scholar] [CrossRef]
- Coque, T.M.; Novais, A.; Carattoli, A.; Poirel, L.; Pitout, J.; Peixe, L.; Baquero, F.; Canton, R.; Nordmann, P. Dissemination of clonally related Escherichia coli strains expressing extended-spectrum β-lactamase CTX-M-15. Emerg. Infect. Dis. 2008, 14, 195–200. [Google Scholar] [CrossRef]
- Boyd, D.A.; Tyler, S.; Christianson, S.; McGeer, A.; Muller, M.P.; Willey, B.M.; Bryce, E.; Gardam, M.; Nordmann, P.; Mulvey, M.R. Complete nucleotide sequence of a 92-kilobase plasmid harboring the CTX-M-15 extended-spectrum beta-lactamase involved in an outbreak in long-term-care facilities in Toronto, Canada. Antimicrob. Agents Chemother. 2004, 48, 3758–3764. [Google Scholar] [CrossRef]
- Feng, Y.; Mannion, A.; Madden, C.M.; Swennes, A.G.; Townes, C.; Byrd, C.; Marini, R.P.; Fox, J.G. Cytotoxic Escherichia coli strains encoding colibactin and cytotoxic necrotizing factor (CNF) colonize laboratory macaques. Gut Pathog. 2017, 9, 71. [Google Scholar] [CrossRef]
- Chaoprasid, P.; Dersch, P. The cytotoxic necrotizing factors (CNFs)—A family of Rho GTPase-activating bacterial exotoxins. Toxins 2021, 13, 901. [Google Scholar] [CrossRef]
- Wang, M.C.; Fan, Y.H.; Zhang, Y.Z.; Bregente, C.J.B.; Lin, W.H.; Chen, C.A.; Lin, T.P.; Kao, C.Y. Characterization of uropathogenic Escherichia coli phylogroups associated with antimicrobial resistance, virulence factor distribution, and virulence-related phenotypes. Infect. Genet. Evol. 2023, 114, 105493. [Google Scholar] [CrossRef]
- Paramita, R.I.; Nelwan, E.J.; Fadilah, F.; Renesteen, E.; Puspandari, N.; Erlina, L. Genome-based characterization of Escherichia coli causing bloodstream infection through next-generation sequencing. PLoS ONE 2020, 15, e0244358. [Google Scholar] [CrossRef]
- Deku, D.G.; Duedu, K.O.; Kinanyok, S.; Kpene, G.E.; Feglo, P.K. Phylogenicity and virulence profiles of clinical Escherichia coli isolates in the Ho Teaching Hospital of Ghana. Biomed. Res. Int. 2022, 2022, 1347033. [Google Scholar] [CrossRef]
- Flament-Simon, S.C.; Nicolas-Chanoine, M.H.; Garcia, V.; Duprilot, M.; Mayer, N.; Alonso, M.P.; Garcia-Menino, I.; Blanco, J.E.; Blanco, M.; Blanco, J. Clonal structure, virulence factor-encoding genes and antibiotic resistance of Escherichia coli, causing urinary tract infections and other extraintestinal infections in humans in Spain and France during 2016. Antibiotics 2020, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Murase, K.; Martin, P.; Porcheron, G.; Houle, S.; Helloin, E.; Penary, M.; Nougayrede, J.P.; Dozois, C.M.; Hayashi, T.; Oswald, E. HlyF produced by extraintestinal pathogenic Escherichia coli is a virulence factor that regulates outer membrane vesicle biogenesis. J. Infect. Dis. 2016, 213, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Gigliucci, F.; van Hoek, A.H.A.M.; Chiani, P.; Knijn, A.; Minelli, F.; Scavia, G.; Franz, E.; Morabito, S.; Michelacci, V. Genomic characterization of hlyF-positive shiga toxin-producing Escherichia coli, Italy and the Netherlands, 2000–2019. Emerg. Infect. Dis. 2021, 27, 853–861. [Google Scholar] [CrossRef]
- Ferreira, M.R.A.; Silva, T.S.; Stella, A.E.; Conceicao, F.R.; dos Reis, E.F.; Moreira, C.N. Detection of virulence factors and antimicrobial resistance patterns in shiga toxin-producing Escherichia coli isolates from sheep. Pesq. Vet. Bras. 2015, 35, 775–780. [Google Scholar] [CrossRef]
- Karakaya, E.; Aydin, F.; Kayman, T.; Abay, S. Escherichia coli in different animal feces: Phylotypes and virulence genes. World J. Microbiol. Biotechnol. 2022, 39, 14. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Y.; Xiang, J.; Xiang, S.; Zhao, Y.; Xiao, M.; Du, F.; Ji, H.; Kaboli, P.J.; Wu, X.; et al. Metagenome analysis of intestinal bacteria in healthy people, patients with inflammatory bowel disease and colorectal cancer. Front. Cell. Infect. Microbiol. 2021, 11, 599734. [Google Scholar] [CrossRef]
- Suez, J.; Zmora, N.; Zilberman-Schapira, G.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Zur, M.; Regev-Lehavi, D.; Ben-Zeev Brik, R.; Federici, S.; et al. Post-antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell 2018, 174, 1406–1423. [Google Scholar] [CrossRef]
- Montassier, E.; Valdes-Mas, R.; Batard, E.; Zmora, N.; Dori-Bachash, M.; Suez, J.; Elinav, E. Probiotics impact the antibiotic resistance gene reservoir along the human GI tract in a person-specific and antibiotic-dependent manner. Nat. Microbiol. 2021, 6, 1043–1054. [Google Scholar] [CrossRef]
- Frolova, M.; Yudin, S.; Makarov, V.; Glazunova, O.; Alikina, O.; Markelova, N.; Kolzhetsov, N.; Dzhelyadin, T.; Shcherbakova, V.; Trubitsyn, V.; et al. Lacticaseibacillus paracasei: Occurrence in the human gut microbiota and k-mer-based assessment of intraspecies diversity. Life 2021, 11, 1246. [Google Scholar] [CrossRef]
- Meslier, V.; Laiola, M.; Roager, H.; De Filippis, F.; Roume, H.; Quinquis, B.; Giacco, R.; Mennella, I.; Ferracane, R.; Pons, N.; et al. Mediterranean diet intervention in overweight and obese subjects lowers plasma cholesterol and causes changes in the gut microbiome and metabolome independently of energy intake. Gut 2020, 69, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Kruger, F. Babraham Bioinformatics—Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 7 April 2025).
- Sorensen, T. A method of establishing groups of equal amplitude in plant sociology based on similarity of species content. Biol. Skr. 1948, 5, 1–34. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar] [CrossRef]
- Mann, H.B.; Whitney, D.R. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- VassarStats: Calculators for Statistical Table Entries r to P. Available online: http://vassarstats.net/tabs_r.html (accessed on 18 April 2025).
- Quenouille, M.H. Approximate tests of correlation in time-series. J. R. Stat. Soc. Ser. B 1949, 11, 68–84. [Google Scholar] [CrossRef]
- Qi, W.; Lacher, D.W.; Bumbaugh, A.C.; Hyma, K.E.; Quellette, L.M.; Large, T.M.; Tarr, C.L.; Whittam, T.S. EcMLST: An online database for multi locus sequence typing of pathogenic Escherichia coli. In Proceedings of the 2004 IEEE Computational Systems Bioinformatics Conference, Stanford, CA, USA, 19 August 2004; pp. 520–521. [Google Scholar] [CrossRef]
- Wirth, T.; Falush, D.; Lan, R.; Colles, F.; Mensa, P.; Wieler, L.H.; Karch, H.; Reeves, P.R.; Maiden, M.C.J.; Ochman, H.; et al. Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 2006, 60, 1136–1151. [Google Scholar] [CrossRef]
- Lescat, M.; Hoede, C.; Clermont, O.; Garry, L.; Darlu, P.; Tuffery, P.; Denamur, E.; Picard, B. aes, the gene encoding the esterase B in Escherichia coli, is a powerful phylogenetic marker of the species. BMC Microbiol. 2009, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Madec, J.-Y.; Lupo, A.; Schink, A.-K.; Kieffer, N.; Nordmann, P.; Schwarz, S. Antimicrobial resistance in Escherichia coli. Microbiol. Spectr. 2018, 6, ARBA-0026-2017. [Google Scholar] [CrossRef]
- Mohsin, A.S.; Alsakini, A.H.; Ali, M.R. Outbreak of drug resistance Escherichia coli phylogenetic F group associated urinary tract infection. Iran. J. Microbiol. 2022, 14, 341–350. [Google Scholar] [CrossRef]
- Neyestani, Z.; Khademi, F.; Teimourpour, R.; Amani, M.; Arzanlou, M. Prevalence and mechanisms of ciprofloxacin resistance in Escherichia coli isolated from hospitalized patients, healthy carriers, and wastewaters in Iran. BMC Microbiol. 2023, 23, 191. [Google Scholar] [CrossRef]
- Onderdonk, A.B.; Louie, T.J.; Tally, F.P.; Bartlett, J.G. Activity of metronidazole against Escherichia coli in experimental infra-abdominal sepsis. J. Antimicrob. Chemother. 1979, 5, 201–210. [Google Scholar] [CrossRef]
- Kolzhetsov, N.; Markelova, N.; Frolova, M.; Alikina, O.; Glazunova, O.; Safonova, L.; Kalashnikova, I.; Yudin, V.; Makarov, V.; Keskinov, A.; et al. Enterotype-dependent probiotic-mediated changes in the male rat intestinal microbiome in vivo and in vitro. Int. J. Mol. Sci. 2024, 25, 4558. [Google Scholar] [CrossRef]
- Andoh, A.; Nishida, A.; Takahashi, K.; Inatomi, O.; Imaeda, H.; Bamba, S.; Kito, K.; Sugimoto, M.; Kobayashi, T. Comparison of the gut microbial community between obese and lean peoples using 16S gene sequencing in a Japanese population. J. Clin. Biochem. Nutr. 2016, 59, 65. [Google Scholar] [CrossRef]
- Shavkunov, K.S.; Markelova, N.Y.; Glazunova, O.A.; Kolzhetsov, N.P.; Panyukov, V.V.; Ozoline, O.N. The Fate and Functionality of Alien tRNA Fragments in Culturing Medium and Cells of Escherichia coli. Int. J. Mol. Sci. 2023, 24, 12960. [Google Scholar] [CrossRef]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef]
- Cuklina, J.; Pedrioli, P.G.A.; Aebersold, R. Review of batch effects prevention, diagnostics, and correction approaches. Methods Mol. Biol. 2020, 2051, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Dubnau, D.; Losick, R. Bistability in bacteria. Mol. Microbiol. 2006, 61, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Smits, W.K.; Kuipers, O.P.; Veening, J.W. Phenotypic variation in bacteria: The role of feedback regulation. Nat. Rev. Microbiol. 2006, 4, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Ronin, I.; Katsowich, N.; Rosenshine, I.; Balaban, N.Q. A long-term epigenetic memory switch controls bacterial virulence bimodality. eLife 2017, 6, e19599. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, S.; Zhang, Y.; Zhang, W. DNA adenine methylation is involved in persister formation in E. coli. Microbiol. Res. 2021, 246, 126709. [Google Scholar] [CrossRef]
- Foster, P.L. Stress-induced mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 373–397. [Google Scholar] [CrossRef]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef]
- Raser, J.M.; O’Shea, E.K. Noise in gene expression: Origins, consequences, and control. Science 2005, 309, 2010–2013. [Google Scholar] [CrossRef]
- Korobkova, E.; Emonet, T.; Vilar, J.M.G.; Shimizu, T.S.; Cluzel, P. From molecular noise to behavioural variability in a single bacterium. Nature 2004, 428, 574–578. [Google Scholar] [CrossRef]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef]
- Elhenawy, W.; Tsai, N.T.; Coombes, B.K. Host-specific adaptive diversification of Crohn’s disease-associated adherent-invasive Escherichia coli. Cell Host Microbe 2019, 25, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Denamur, E.; Clermont, O.; Bonacorsi, S.; Gordon, D. The population genetics of pathogenic Escherichia coli. Nat. Rev. Microbiol. 2021, 19, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Mirsepasi-Lauridsen, H.C.; Vallance, B.A.; Krogfelt, K.A.; Petersen, A.M. Escherichia coli pathobionts associated with inflammatory bowel disease. Clin. Microbiol. Rev. 2019, 30, e00060-18. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 29, 623–628. [Google Scholar] [CrossRef]
- Wexler, A.G.; Goodman, A.L. An insider’s perspective: Bacteroides as a window into the microbiome. Nat. Microbiol. 2017, 2, 17026. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Dataset | Donor Types and Number of Samples | Bioproject | |
|---|---|---|---|
| Types | Number | ||
| Inflammatory bowel diseases | Healthy individuals | 65 | PRJEB7774 [48] |
| Patients with adenoma | 49 | ||
| Patients with carcinoma | 46 | ||
| Antibiotic treatment with or without probiotic recovery | Fifteen healthy donors before antibiotic treatment | 92 | PRJEB28097 [49,50] |
| Twelve donors of the same group during antibiotic treatment | 49 | ||
| Seven donors of the same group during self-recovery | 42 | ||
| Eight donors of the same goup during recovery with probiotics | 43 | ||
| Overweight donors before and after diet | Samples from 43 overweight or obese individuals | 86 | PRJEB33500 [52] |
| Samples from the same 43 persons after Mediterranean diet | 86 | ||
| Phylogroups | Number of Genomes | Number of 18-Mers | ||
|---|---|---|---|---|
| Set 1 | Set 2 | Set 1 | Set 2 | |
| A | 17 | 21 | 415,335 | 354,997 |
| B1 | 25 | 25 | 710,784 | 524,927 |
| B2 | 23 | 29 | 1,014,716 | 783,899 |
| C | 14 | 17 | 242,272 | 170,224 |
| D | 11 | 15 | 673,338 | 524,936 |
| E | 13 | 19 | 680,604 | 802,163 |
| F | 11 | 15 | 445,835 | 313,383 |
| G | 10 | 13 | 254,624 | 171,176 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frolova, M.S.; Kiselev, S.S.; Panyukov, V.V.; Ozoline, O.N. Phylogroup Homeostasis of Escherichia coli in the Human Gut Reflects the Physiological State of the Host. Microorganisms 2025, 13, 1584. https://doi.org/10.3390/microorganisms13071584
Frolova MS, Kiselev SS, Panyukov VV, Ozoline ON. Phylogroup Homeostasis of Escherichia coli in the Human Gut Reflects the Physiological State of the Host. Microorganisms. 2025; 13(7):1584. https://doi.org/10.3390/microorganisms13071584
Chicago/Turabian StyleFrolova, Maria S., Sergey S. Kiselev, Valery V. Panyukov, and Olga N. Ozoline. 2025. "Phylogroup Homeostasis of Escherichia coli in the Human Gut Reflects the Physiological State of the Host" Microorganisms 13, no. 7: 1584. https://doi.org/10.3390/microorganisms13071584
APA StyleFrolova, M. S., Kiselev, S. S., Panyukov, V. V., & Ozoline, O. N. (2025). Phylogroup Homeostasis of Escherichia coli in the Human Gut Reflects the Physiological State of the Host. Microorganisms, 13(7), 1584. https://doi.org/10.3390/microorganisms13071584