
Development of a TaqMan One-Step Quantitative PCR Assay for the Simultaneous Detection of Novel Goose Parvovirus and Novel Duck Reovirus
 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
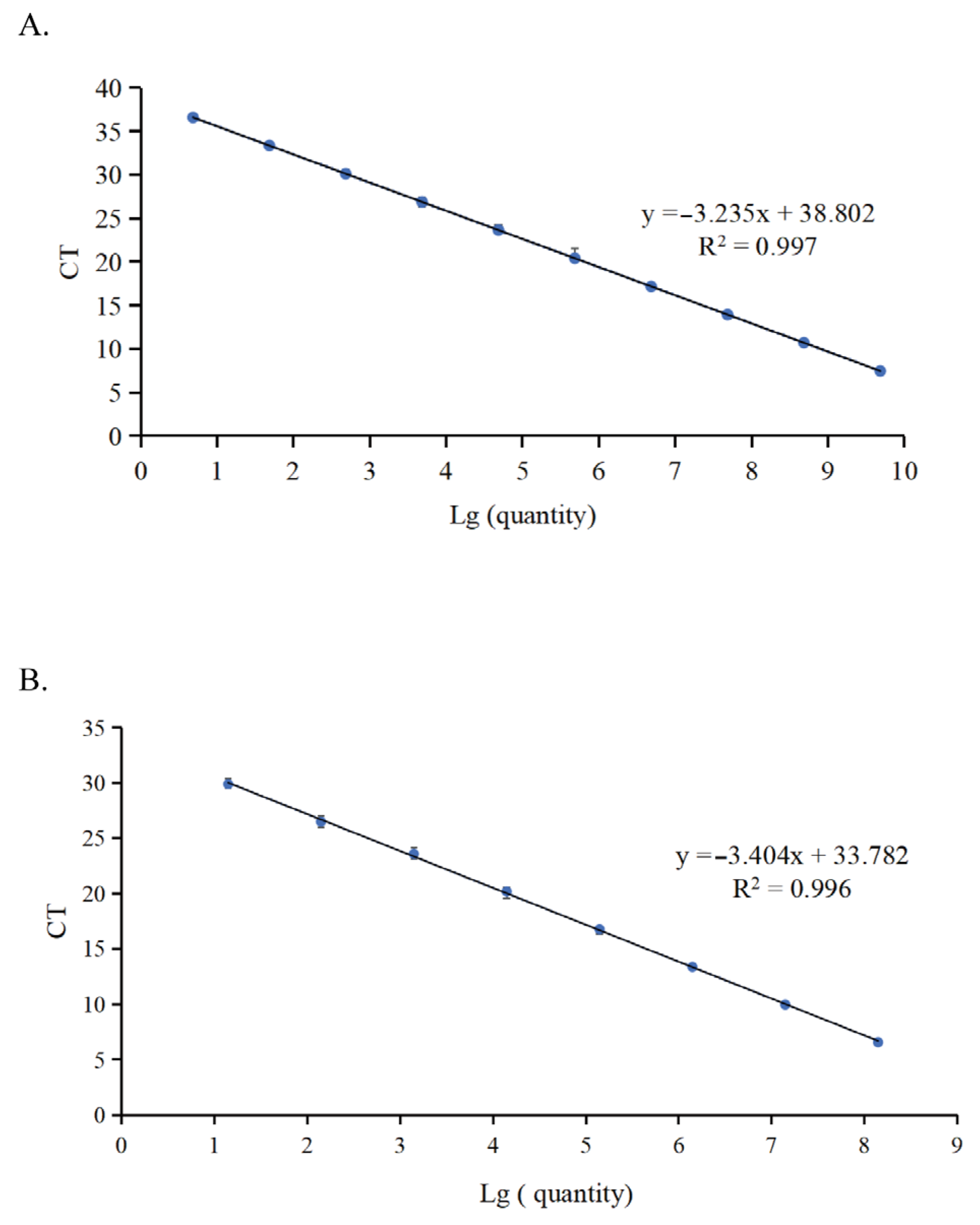
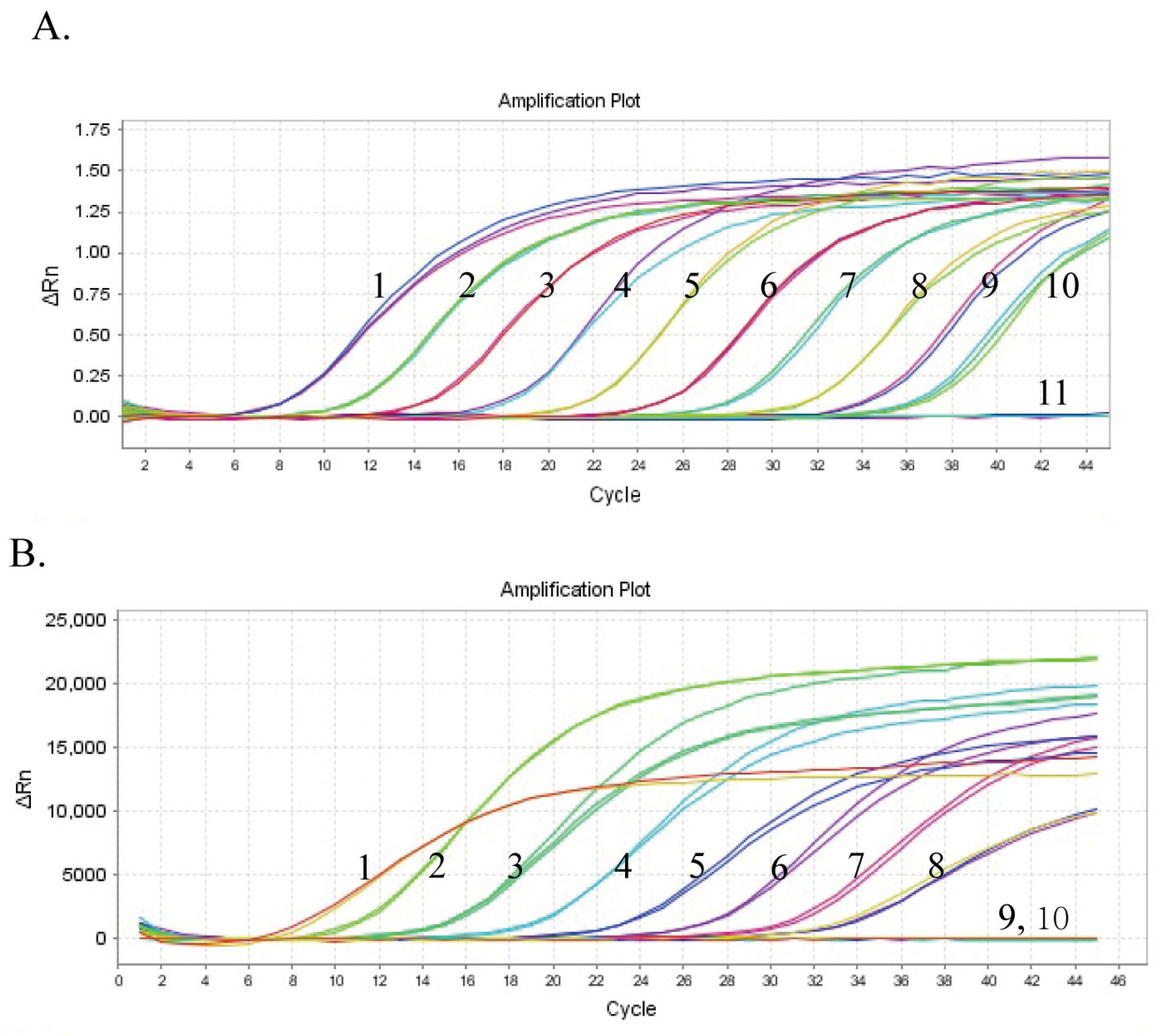
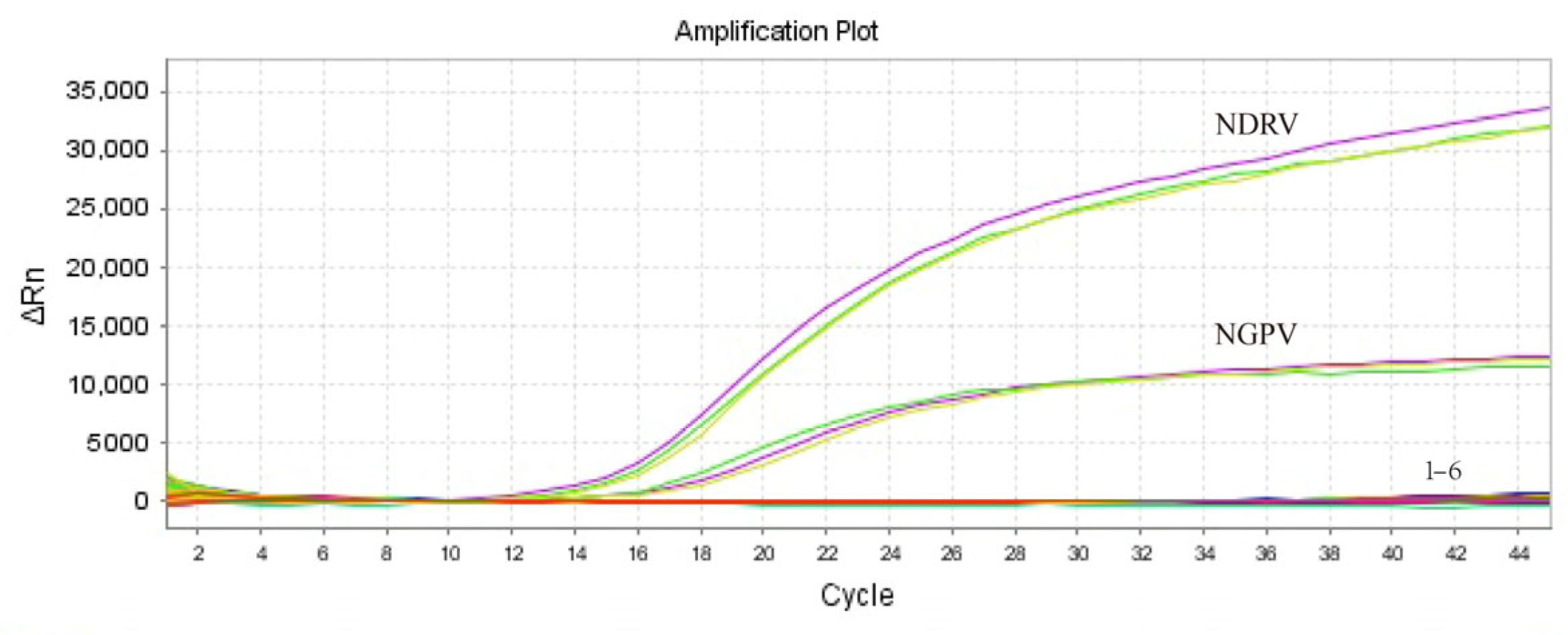
3. Results and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, S.; Wang, S.; Cheng, X.; Xiao, S.; Zhu, X.; Lin, F.; Wu, N.; Wang, J.; Huang, M.; Zheng, M.; et al. Isolation and characterization of a distinct duck-origin goose parvovirus causing an outbreak of duckling short beak and dwarfism syndrome in China. Arch. Virol. 2016, 161, 2407–2416. [Google Scholar] [CrossRef]
- Yu, K.; Ma, X.; Sheng, Z.; Qi, L.; Liu, C.; Wang, D.; Huang, B.; Li, F.; Song, M. Identification of Goose-Origin Parvovirus as a Cause of Newly Emerging Beak Atrophy and Dwarfism Syndrome in Ducklings. J. Clin. Microbiol. 2016, 54, 1999–2007. [Google Scholar] [CrossRef]
- Chen, Y.; Afumba, R.; Pang, F.; Yuan, R.; Dong, H. Advances in Research on Genetic Relationships of Waterfowl Parvoviruses. J. Vet. Res. 2021, 65, 391–399. [Google Scholar] [CrossRef]
- Huo, X.; Chen, Y.; Zhu, J.; Wang, Y. Evolution, genetic recombination, and phylogeography of goose parvovirus. Comp. Immunol. Microbiol. Infect. Dis. 2023, 102, 102079. [Google Scholar] [CrossRef]
- Huong, N.T.; Hieu, D.V.; Bich, N.T.; Khanh, T.V.; Ba, N.T.; Xuan, C.T.N.; Hien, Q.T.M.; Thai, T.H.; Huong, C.T.T. Isolation and genetic characterization of waterfowl parvovirus in ducks in Northern Vietnam. Vet. World 2024, 17, 981–987. [Google Scholar] [CrossRef]
- Matczuk, A.K.; Chmielewska-Władyka, M.; Siedlecka, M.; Bednarek, K.J.; Wieliczko, A. Short Beak and Dwarfism Syndrome in Ducks in Poland Caused by Novel Goose Parvovirus. Animals 2020, 10, 2397. [Google Scholar] [CrossRef]
- Rosenberger, J.K.; Sterner, F.J.; Botts, S.; Lee, K.P.; Margolin, A. In vitro and in vivo characterization of avian reoviruses. I. Pathogenicity and antigenic relatedness of several avian reovirus isolates. Avian Dis. 1989, 33, 535–544. [Google Scholar] [CrossRef]
- Kong, J.; Shao, G.; Zhang, Y.; Wang, J.; Xie, Z.; Feng, K.; Zhang, X.; Xie, Q. Molecular characterization, complete genome sequencing, and pathogenicity of Novel Duck Reovirus from South Coastal Area in China. Poult. Sci. 2023, 102, 102776. [Google Scholar] [CrossRef]
- Li, N.; Hong, T.; Wang, Y.; Wang, Y.; Yu, K.; Cai, Y.; Liu, S.; Wei, L.; Chai, T. The pathogenicity of novel duck reovirus in Cherry Valley ducks. Vet. Microbiol. 2016, 192, 181–185. [Google Scholar] [CrossRef]
- Wang, H.; Gao, B.; Liu, X.; Zhang, S.; Diao, Y.; Tang, Y. Pathogenicity of a variant duck orthoreovirus strain in Cherry Valley Ducklings. Vet. Microbiol. 2020, 242, 108546. [Google Scholar] [CrossRef]
- Contreras, N.E.; Roldán, J.S.; Castillo, D.S. Novel competitive enzyme-linked immunosorbent assay for the detection of the high-risk Human Papillomavirus 18 E6 oncoprotein. PLoS ONE 2023, 18, e0290088. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Huang, Y.; Chen, G.; Shi, M.; Qiao, Y.; Huang, T.; Wei, T.; Mo, M.; He, X.; et al. Rapid and visual detection of the emerging novel duck reovirus by using a specific and sensitive reverse transcription recombinase polymerase amplification method. J. Virol. Methods 2021, 291, 114098. [Google Scholar] [CrossRef]
- Yang, J.; Chen, H.; Wang, Z.; Yu, X.; Niu, X.; Tang, Y.; Diao, Y. Development of a Quantitative Loop-Mediated Isothermal Amplification Assay for the Rapid Detection of Novel Goose Parvovirus. Front. Microbiol. 2017, 8, 2472. [Google Scholar] [CrossRef]
- Yun, T.; Hua, J.; Ye, W.; Chen, L.; Ni, Z.; Zhu, Y.; Zhang, C. Development and Evaluation of a Monoclonal Antibody-Based Blocking Enzyme-Linked Immunosorbent Assay for the Detection of Antibodies against Novel Duck Reovirus in Waterfowl Species. Microbiol. Spectr. 2022, 10, e0258122. [Google Scholar] [CrossRef]
- Kim, J.; Easley, C.J. Isothermal DNA amplification in bioanalysis: Strategies and applications. Bioanalysis 2011, 3, 227–239. [Google Scholar] [CrossRef]
- Liu, W.J.; Yang, Y.T.; Du, S.M.; Yi, H.D.; Xu, D.N.; Cao, N.; Jiang, D.L.; Huang, Y.M.; Tian, Y.B. Rapid and sensitive detection of goose parvovirus and duck-origin novel goose parvovirus by recombinase polymerase amplification combined with a vertical flow visualization strip. J. Virol. Methods 2019, 266, 34–40. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, G.; Ding, X.; Zhang, K.; Sun, W.; Li, Q.; Yi, Y.; Wang, J.; Pang, X.; Chen, L. A rapid simultaneous detection of duck hepatitis A virus 3 and novel duck reovirus based on RPA CRISPR Cas12a/Cas13a. Int. J. Biol. Macromol. 2024, 274, 133246. [Google Scholar] [CrossRef]
- Niu, X.; Chen, H.; Yang, J.; Yu, X.; Ti, J.; Wang, A.; Diao, Y. Development of a TaqMan-based real-time PCR assay for the detection of Novel GPV. J. Virol. Methods 2016, 237, 32–37. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Cui, Y.; Nan, H.; Yuan, W. Development of a TaqMan-based real-time PCR assay for the rapid and specific detection of novel duck- origin goose parvovirus. Mol. Cell. Probes 2017, 34, 56–58. [Google Scholar] [CrossRef]
- Zhang, S.; Li, W.; Liu, X.; Li, X.; Gao, B.; Diao, Y.; Tang, Y. A TaqMan-based real-time PCR assay for specific detection of novel duck reovirus in China. BMC Vet. Res. 2020, 16, 306. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Bi, Z.; Liu, Y.; Meng, C.; Zhu, J.; Liu, G.; Li, C. Simultaneous detection of novel goose parvovirus and novel duck reovirus by SYBR Green I-based duplex real-time quantitative polymerase chain reaction. 3 Biotech 2024, 14, 288. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Category | Concentration (Copies/μL) | Mean Ct | SD | CV (%) |
|---|---|---|---|---|
| Intra-assay | 104 | 24.130 | 0.074 | 0.32 |
| 105 | 20.919 | 0.052 | 0.26 | |
| 106 | 17.116 | 0.024 | 0.14 | |
| Inter-assay | 104 | 24.863 | 0.043 | 0.17 |
| 105 | 20.981 | 0.055 | 0.26 | |
| 106 | 17.319 | 0.108 | 0.62 |
| Category | Concentration (Copies/μL) | Mean Ct | SD | CV (%) |
|---|---|---|---|---|
| Intra-assay | 104 | 20.510 | 0.057 | 0.27 |
| 105 | 16.847 | 0.144 | 0.85 | |
| 106 | 13.060 | 0.072 | 0.55 | |
| Inter-assay | 104 | 20.110 | 0.160 | 0.79 |
| 105 | 16.391 | 0.073 | 0.44 | |
| 106 | 12.840 | 0.136 | 1.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, Y.; Bi, Z.; Wang, J.; Wang, G.; Ru, X.; Meng, C.; Zhu, J.; Liu, G.; Li, C. Development of a TaqMan One-Step Quantitative PCR Assay for the Simultaneous Detection of Novel Goose Parvovirus and Novel Duck Reovirus. Microorganisms 2025, 13, 1582. https://doi.org/10.3390/microorganisms13071582
Wang Y, Wang Y, Bi Z, Wang J, Wang G, Ru X, Meng C, Zhu J, Liu G, Li C. Development of a TaqMan One-Step Quantitative PCR Assay for the Simultaneous Detection of Novel Goose Parvovirus and Novel Duck Reovirus. Microorganisms. 2025; 13(7):1582. https://doi.org/10.3390/microorganisms13071582
Chicago/Turabian StyleWang, Yimin, Yong Wang, Zhuangli Bi, Jinbin Wang, Gang Wang, Xin Ru, Chunchun Meng, Jie Zhu, Guangqing Liu, and Chuanfeng Li. 2025. "Development of a TaqMan One-Step Quantitative PCR Assay for the Simultaneous Detection of Novel Goose Parvovirus and Novel Duck Reovirus" Microorganisms 13, no. 7: 1582. https://doi.org/10.3390/microorganisms13071582
APA StyleWang, Y., Wang, Y., Bi, Z., Wang, J., Wang, G., Ru, X., Meng, C., Zhu, J., Liu, G., & Li, C. (2025). Development of a TaqMan One-Step Quantitative PCR Assay for the Simultaneous Detection of Novel Goose Parvovirus and Novel Duck Reovirus. Microorganisms, 13(7), 1582. https://doi.org/10.3390/microorganisms13071582