
Impact of SARS-CoV-2 P.1 Variant Infection on the Nasopharyngeal Commensal Bacterial Microbiome of Individuals from the Brazilian Amazon

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Study Population and Sample Collection
2.3. Tests to Detect the Genome of SARS-CoV-2 and Other Respiratory Viruses
2.4. Shotgun Metagenomic Next-Generation Sequencing
2.5. Bioinformatics Analysis
2.6. Statistical Analysis
3. Results
3.1. Sample Characteristics and Study Population
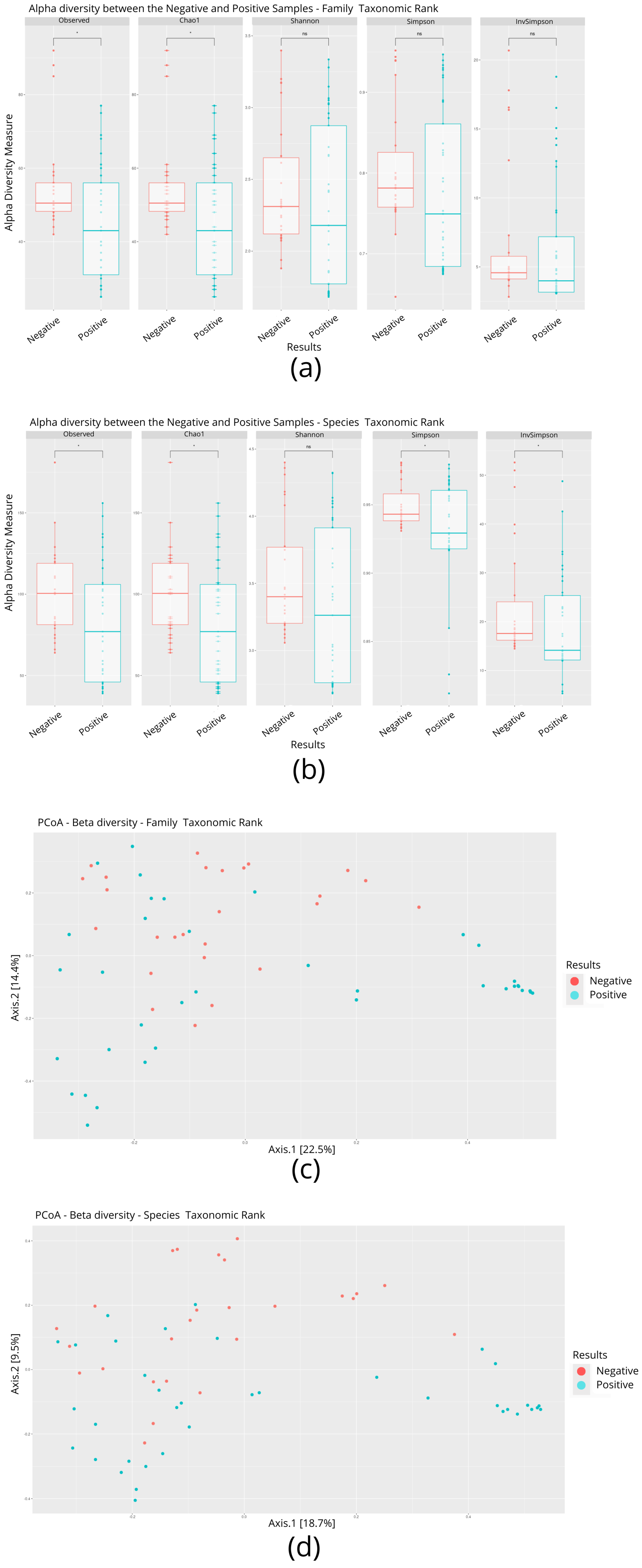
3.2. Microbial Diversity of Individuals Infected and Not Infected with SARS-CoV-2
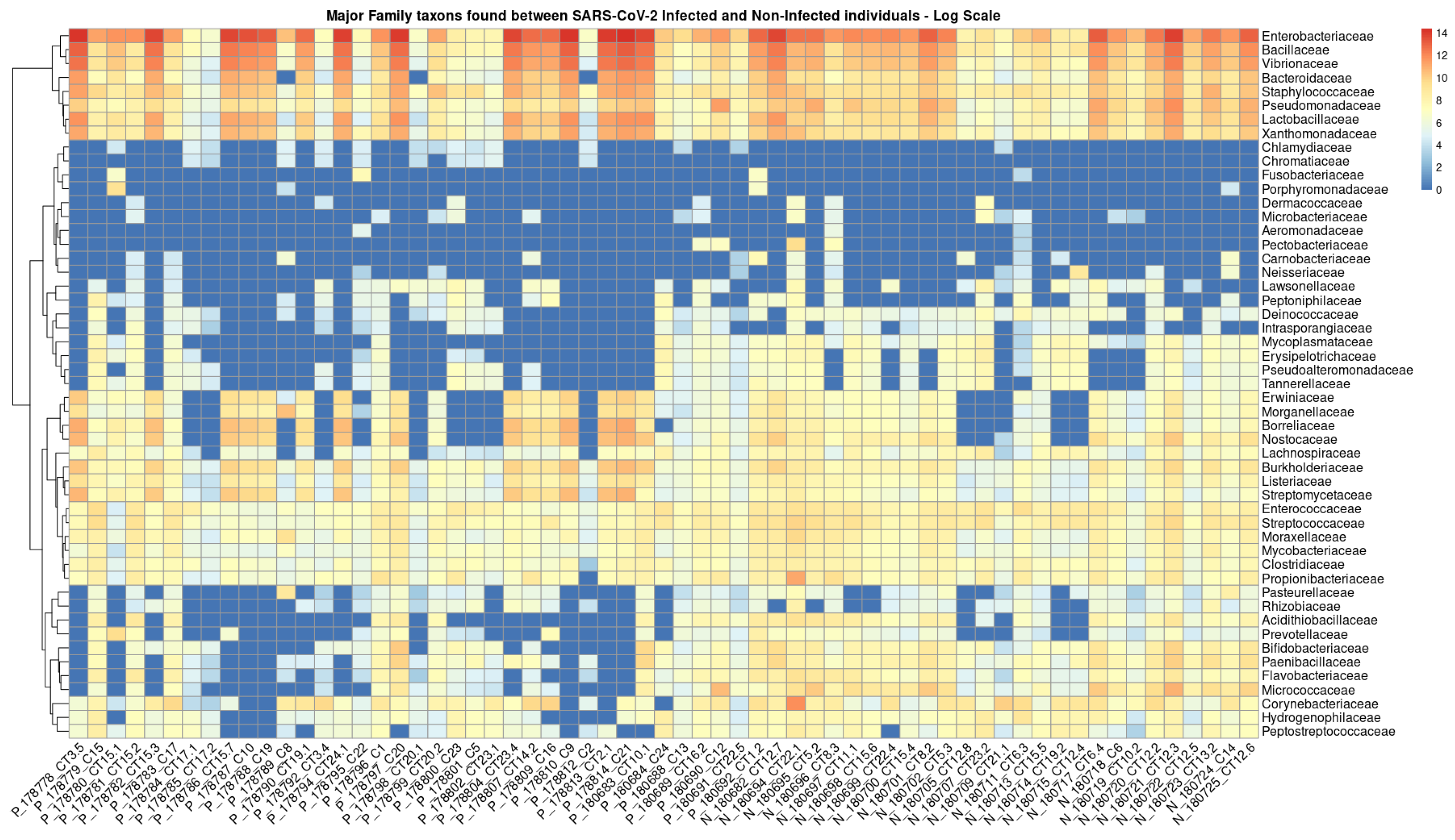
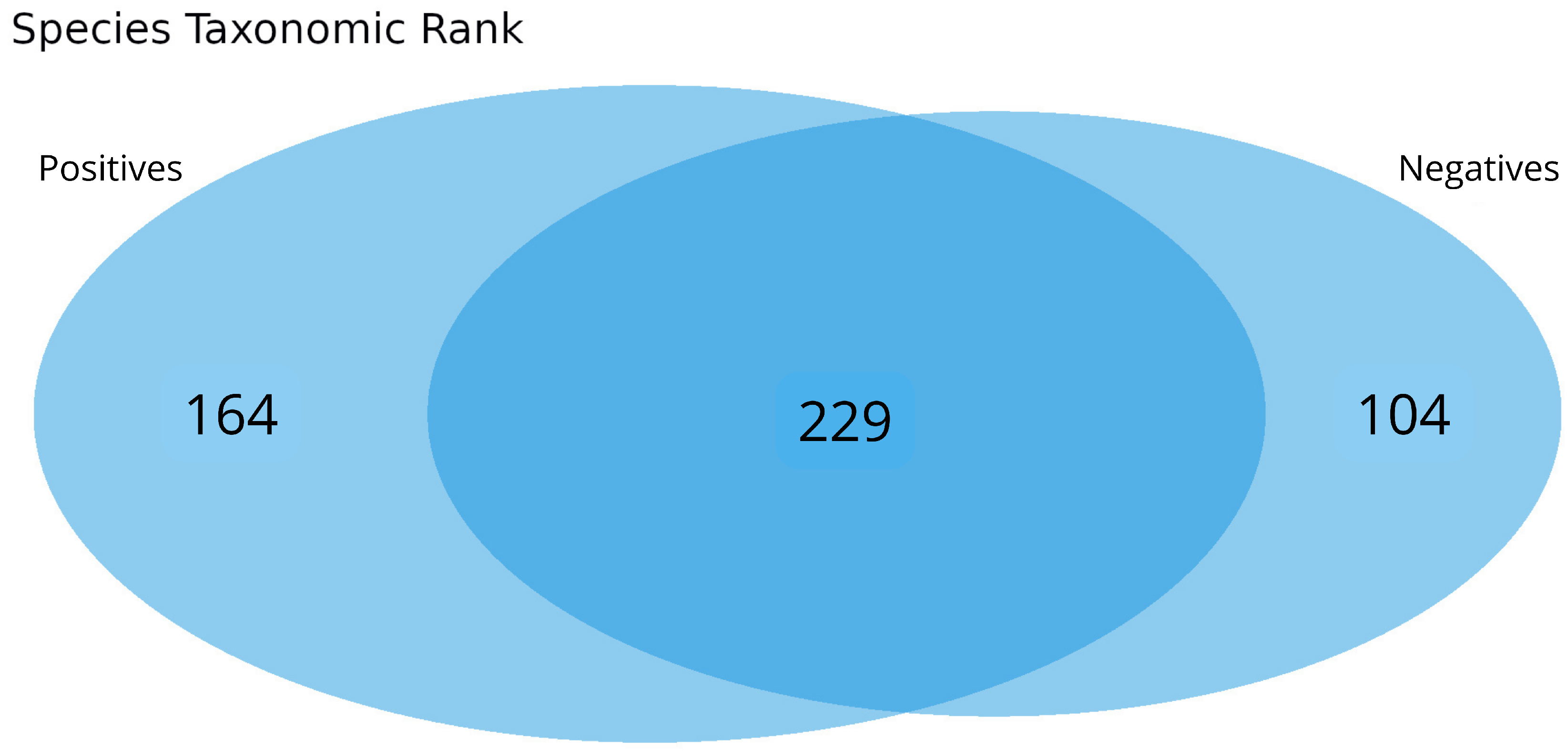
3.3. Intra-Family and Inter-Family Microbial Profile
3.4. Bacterial Communities Associated with Age Groups and Sex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.P.; Day, T.; Arino, J.; Colijn, C.; Dushoff, J.; Li, M.; Mechai, S.; Van Domselaar, G.; Wu, J.; Earn, D.J.; et al. The origins and potential future of SARS-CoV-2 variants of concern in the evolving COVID-19 pandemic. Curr. Biol. 2021, 31, R918–R929. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, W. WHO-Convened Global Study of Origins of SARS-CoV-2: China Part (Text Extract). Infect. Dis. Immun. 2021, 1, 125–132. [Google Scholar] [CrossRef]
- Yamamoto, S.; Saito, M.; Tamura, A.; Prawisuda, D.; Mizutani, T.; Yotsuyanagi, H. The human microbiome and COVID-19: A systematic review. PLoS ONE 2021, 16, e0253293. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Annu. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef]
- Cleary, D.W.; Clarke, S.C. The nasopharyngeal microbiome. Emerg. Top. Life Sci. 2017, 1, 297–312. [Google Scholar] [CrossRef]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef]
- Li, N.; Ma, W.T.; Pang, M.; Fan, Q.L.; Hua, J.L. The commensal microbiota and viral infection: A comprehensive review. Front. Immunol. 2019, 10, 1551. [Google Scholar] [CrossRef]
- Man, W.H.; de Steenhuijsen Piters, W.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef]
- Rosas-Salazar, C.; Kimura, K.S.; Shilts, M.H.; Strickland, B.A.; Freeman, M.H.; Wessinger, B.C.; Gupta, V.; Brown, H.M.; Rajagopala, S.V.; Turner, J.H.; et al. SARS-CoV-2 infection and viral load are associated with the upper respiratory tract microbiome. J. Allergy Clin. Immunol. 2021, 147, 1226–1233.e2. [Google Scholar] [CrossRef] [PubMed]
- Edouard, S.; Million, M.; Bachar, D.; Dubourg, G.; Michelle, C.; Ninove, L.; Charrel, R.; Raoult, D. The nasopharyngeal microbiota in patients with viral respiratory tract infections is enriched in bacterial pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, L.; Charitos, I.A.; Ballini, A.; Inchingolo, F.; Luperto, P.; De Nitto, E.; Topi, S. The human respiratory system and its microbiome at a glimpse. Biology 2020, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Ventero, M.P.; Cuadrat, R.R.C.; Vidal, I.; Andrade, B.G.N.; Molina-Pardines, C.; Haro-Moreno, J.M.; Coutinho, F.H.; Merino, E.; Regitano, L.C.A.; Silveira, C.B.; et al. Nasopharyngeal Microbial Communities of Patients Infected With SARS-CoV-2 That Developed COVID-19. Front. Microbiol. 2021, 12, 637430. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, F.; Zhou, F.; Li, H.; Ge, W.; Gan, R.; Nie, H.; Li, B.; Wang, Y.; Wu, M.; et al. Metagenomic analysis reveals oropharyngeal microbiota alterations in patients with COVID-19. Signal Transduct. Target. Ther. 2021, 6, 191. [Google Scholar] [CrossRef]
- Belvoncikova, P.; Splichalova, P.; Videnska, P.; Gardlik, R. The Human Mycobiome: Colonization, Composition and the Role in Health and Disease. J. Fungi 2022, 8, 1046. [Google Scholar] [CrossRef]
- Candel, S.; Tyrkalska, S.D.; Álvarez-Santacruz, C.; Mulero, V. The nasopharyngeal microbiome in COVID-19. Emerg. Microbes Infect. 2023, 12, e2165970. [Google Scholar] [CrossRef]
- Gao, Z.; Yu, L.; Cao, L.; Yang, M.; Li, Y.; Lan, Y.; Tang, R.; Huang, Y.; Luan, G.; Liu, Y.; et al. Analysis of coexisting pathogens in nasopharyngeal swabs from COVID-19. Front. Cell. Infect. Microbiol. 2023, 13, 1140548. [Google Scholar] [CrossRef]
- Hoque, M.N.; Sarkar, M.H.; Rahman, M.S.; Akter, S.; Banu, T.A.; Goswami, B.; Jahan, I.; Hossain, M.S.; Shamsuzzaman, A.K.M.; Nafisa, T.; et al. SARS-CoV-2 infection reduces human nasopharyngeal commensal microbiome with inclusion of pathobionts. Sci. Rep. 2021, 11, 24042. [Google Scholar] [CrossRef]
- De Maio, F.; Posteraro, B.; Ponziani, F.R.; Cattani, P.; Gasbarrini, A.; Sanguinetti, M. Nasopharyngeal Microbiota Profiling of SARS-CoV-2 Infected Patients. Biol. Proced. Online 2020, 22, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.K.C.; Cheung, M.K.; Lui, G.C.Y.; Ling, L.; Chan, J.Y.K.; Ng, R.W.Y.; Chan, H.C.; Yeung, A.C.M.; Ho, W.C.S.; Boon, S.S.; et al. Limited Impact of SARS-CoV-2 on the Human Naso-Oropharyngeal Microbiota in Hospitalized Patients. Microbiol. Spectr. 2022, 10, e0219622. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Training Course in Real-Time RT-PCR Assays for Non-Influenza Respiratory Viruses; Instituto Adolfo Lutz: São Paulo, Brazil, 2013. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 5 February 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bağcı, C.; Patz, S.; Huson, D.H. DIAMOND+MEGAN: Fast and Easy Taxonomic and Functional Analysis of Short and Long Microbiome Sequences. Curr. Protoc. 2021, 1, e59. [Google Scholar] [CrossRef]
- Kerfeld, C.A.; Scott, K.M. Using BLAST to teach “E-value-tionary” concepts. PLoS Biol. 2011, 9, e1001014. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; ISBN 3-900051-07-0. Available online: https://www.r-project.org/ (accessed on 5 February 2024).
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Wickham, H.; Chang, W.; Henry, L.; Pedersen, T.L.; Takahashi, K.; Wilke, C.; Woo, K.; Yutani, H.; Dunnington, D.; Posit, P.B.C. ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics; CRAN: Contributed Packages: Vienna, Austria, 2007. [Google Scholar] [CrossRef]
- Li, W.; Ma, Z. The Upper Respiratory Tract Microbiome Network Impacted by SARS-CoV-2. Microb. Ecol. 2023, 86, 1428–1437. [Google Scholar] [CrossRef]
- Gupta, A.; Bhanushali, S.; Karyakarte, R.; Joshi, S.; Das, R.; Shouche, Y.; Sharma, A. Mycobiome profiling of nasopharyngeal region of SARS-CoV-2 infected individuals. Microbes Infect. 2023, 25, 105059. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Song, T.; Zhou, B.; Geber, A.; Ma, Y.; Zhang, L.; Volk, M.; Kapadia, S.N.; Jenkins, S.G.; Salvatore, M.; et al. Microbial Composition of the Human Nasopharynx Varies According to Influenza Virus Type and Vaccination Status. mBio 2019, 10, e01296-19. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Karyakarte, R.; Joshi, S.; Das, R.; Jani, K.; Shouche, Y.; Sharma, A. Nasopharyngeal microbiome reveals the prevalence of opportunistic pathogens in SARS-CoV-2 infected individuals and their association with host types. Microbes Infect. 2022, 24, 104880. [Google Scholar] [CrossRef] [PubMed]
- Sharifipour, E.; Shams, S.; Esmkhani, M.; Khodadadi, J.; Fotouhi-Ardakani, R.; Koohpaei, A.; Doosti, Z.; EJ Golzari, S. Evaluation of bacterial co-infections of the respiratory tract in COVID-19 patients admitted to ICU. BMC Infect. Dis. 2020, 20, 646. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955.e8. [Google Scholar] [CrossRef]
- Devi, P.; Maurya, R.; Mehta, P.; Shamim, U.; Yadav, A.; Chattopadhyay, P.; Kanakan, A.; Khare, K.; Vasudevan, J.S.; Sahni, S.; et al. Increased Abundance of Achromobacter xylosoxidans and Bacillus cereus in Upper Airway Transcriptionally Active Microbiome of COVID-19 Mortality Patients Indicates Role of Co-Infections in Disease Severity and Outcome. Microbiol. Spectr. 2022, 10, e0231121. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, L.; Wang, Y.; Dai, T.; Qin, Z.; Zhou, F.; Zhang, L. Alterations in microbiota of patients with COVID-19: Potential mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 143. [Google Scholar] [CrossRef]
- Xu, R.; Lu, R.; Zhang, T.; Wu, Q.; Cai, W.; Han, X.; Wan, Z.; Jin, X.; Zhang, Z.; Zhang, C. Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Commun. Biol. 2021, 4, 240. [Google Scholar] [CrossRef]
- Gradisteanu Pircalabioru, G.; Grigore, G.A.; Czobor Barbu, I.; Chifiriuc, M.-C.; Savu, O. Impact of COVID-19 on the Microbiome and Inflammatory Status of Type 2 Diabetes Patients. Biomedicines 2023, 11, 179. [Google Scholar] [CrossRef]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Physiol. Behav. 2016, 176, 139–148. [Google Scholar] [CrossRef]
- Alam, S.; Sadiqi, S.; Sabir, M.; Nisa, S.; Ahmad, S.; Abbasi, S.W. Bacillus species; a potential source of anti-SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2022, 40, 5748–5758. [Google Scholar] [CrossRef] [PubMed]
- Du, R.-H.; Liang, L.-R.; Yang, C.-Q.; Wang, W.; Cao, T.-Z.; Li, M.; Guo, G.-Y.; Du, J.; Zheng, C.-L.; Zhu, Q.; et al. Predictors of mortality for patients with COVID-19 pneumonia caused by SARSCoV-2: A prospective cohort study. Eur. Respir. J. 2020, 55, 2000524. [Google Scholar] [CrossRef]
- Suskun, C.; Kilic, O.; Ciftdogan, D.Y.; Guven, S.; Karbuz, A.; Parlakay, A.O.; Kara, Y.; Kacmaz, E.; Sahin, A.; Boga, A.; et al. Intestinal microbiota composition of children with infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and multisystem inflammatory syndrome (MIS-C). Eur. J. Pediatr. 2022, 181, 3175–3191. [Google Scholar] [CrossRef] [PubMed]
- Mahdhi, A.; Hmila, Z.; Behi, A.; Bakhrouf, A. Preliminary characterization of the probiotic properties of candida famata and geobacillus thermoleovorans. Iran. J. Microbiol. 2011, 3, 129–134. [Google Scholar] [PubMed]
- Hughes, S.; Troise, O.; Donaldson, H.; Mughal, N.; Moore, L.S.P. Bacterial and fungal coinfection among hospitalized patients with COVID-19: A retrospective cohort study in a UK secondary-care setting. Clin. Microbiol. Infect. 2020, 26, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Manohar, P.; Loh, B.; Nachimuthu, R.; Hua, X.; Welburn, S.C.; Leptihn, S. Secondary Bacterial Infections in Patients With Viral Pneumonia. Front. Med. 2020, 7, 2013–2016. [Google Scholar] [CrossRef]
- Bielanski, A.; Haber, J. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2013, 486, 330–335. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Kıymet, E.; Böncüoğlu, E.; Şahinkaya, Ş.; Cem, E.; Çelebi, M.Y.; Düzgöl, M.; Kara, A.A.; Arıkan, K.Ö.; Aydın, T.; İşgüder, R.; et al. Distribution of spreading viruses during COVID-19 pandemic: Effect of mitigation strategies. Am. J. Infect. Control 2021, 49, 1142–1145. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kanji, J.N.; Zelyas, N.; Pabbaraju, K.; Granger, D.; Wong, A.; Murphy, S.A.; Buss, E.; MacDonald, C.; Berenger, B.M.; Diggle, M.A.; et al. Respiratory virus coinfections with severe acute respiratory coronavirus virus 2 (SARS-CoV-2) continue to be rare one year into the coronavirus disease 2019 (COVID-19) pandemic in Alberta, Canada (June 2020-May 2021). Infect. Control Hosp. Epidemiol. 2023, 44, 805–808. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. Samples | |
|---|---|
| Variables | n = 63 |
| Individuals infected with SARS-CoV-2 | n = 37 |
| Individuals not infected with SARS-CoV-2 | n = 26 |
| Male | n = 29 |
| Infected | n = 20 |
| Not infected | n = 9 |
| Female | n = 34 |
| Infected | n = 17 |
| Not infected | n = 17 |
| Total Residences | n = 21 |
| Age group 1 (18 to 31 years) | n = 26 |
| Age group 2 (32 to 45 years) | n = 21 |
| Age group 3 (46 to 59 years) | n = 9 |
| Age group 4 (≥60 yeas) | n = 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes Silva Cruz, A.; Cardoso, J.F.; Pinheiro, K.C.; Ferreira, J.A.; Barbagelata, L.S.; Silva, S.P.; Chagas Junior, W.D.; Lobo, P.S.; Teixeira, D.M.; André Junior, W.; et al. Impact of SARS-CoV-2 P.1 Variant Infection on the Nasopharyngeal Commensal Bacterial Microbiome of Individuals from the Brazilian Amazon. Microorganisms 2025, 13, 1088. https://doi.org/10.3390/microorganisms13051088
Mendes Silva Cruz A, Cardoso JF, Pinheiro KC, Ferreira JA, Barbagelata LS, Silva SP, Chagas Junior WD, Lobo PS, Teixeira DM, André Junior W, et al. Impact of SARS-CoV-2 P.1 Variant Infection on the Nasopharyngeal Commensal Bacterial Microbiome of Individuals from the Brazilian Amazon. Microorganisms. 2025; 13(5):1088. https://doi.org/10.3390/microorganisms13051088
Chicago/Turabian StyleMendes Silva Cruz, Amanda, Jedson Ferreira Cardoso, Kenny Costa Pinheiro, Jessylene Almeida Ferreira, Luana Soares Barbagelata, Sandro Patroca Silva, Wanderley Dias Chagas Junior, Patrícia Santos Lobo, Dielle Monteiro Teixeira, Walter André Junior, and et al. 2025. "Impact of SARS-CoV-2 P.1 Variant Infection on the Nasopharyngeal Commensal Bacterial Microbiome of Individuals from the Brazilian Amazon" Microorganisms 13, no. 5: 1088. https://doi.org/10.3390/microorganisms13051088
APA StyleMendes Silva Cruz, A., Cardoso, J. F., Pinheiro, K. C., Ferreira, J. A., Barbagelata, L. S., Silva, S. P., Chagas Junior, W. D., Lobo, P. S., Teixeira, D. M., André Junior, W., Ordenes Silva, I., Santos, M. C., Soares Farias, L. S., Sousa, M. S., & Neto Tavares, F. (2025). Impact of SARS-CoV-2 P.1 Variant Infection on the Nasopharyngeal Commensal Bacterial Microbiome of Individuals from the Brazilian Amazon. Microorganisms, 13(5), 1088. https://doi.org/10.3390/microorganisms13051088