
Genome Mining Reveals Rifamycin Biosynthesis in a Taklamakan Desert Actinomycete

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequence Acquisition and Analysis
2.2. Strain, Plasmid, and Culture Conditions
2.3. Construction of melC-Based Reporter Strains and the LuxR Overexpression Mutant Strain
2.4. Fermentation, Isolation, and Analysis of Metabolites
3. Results
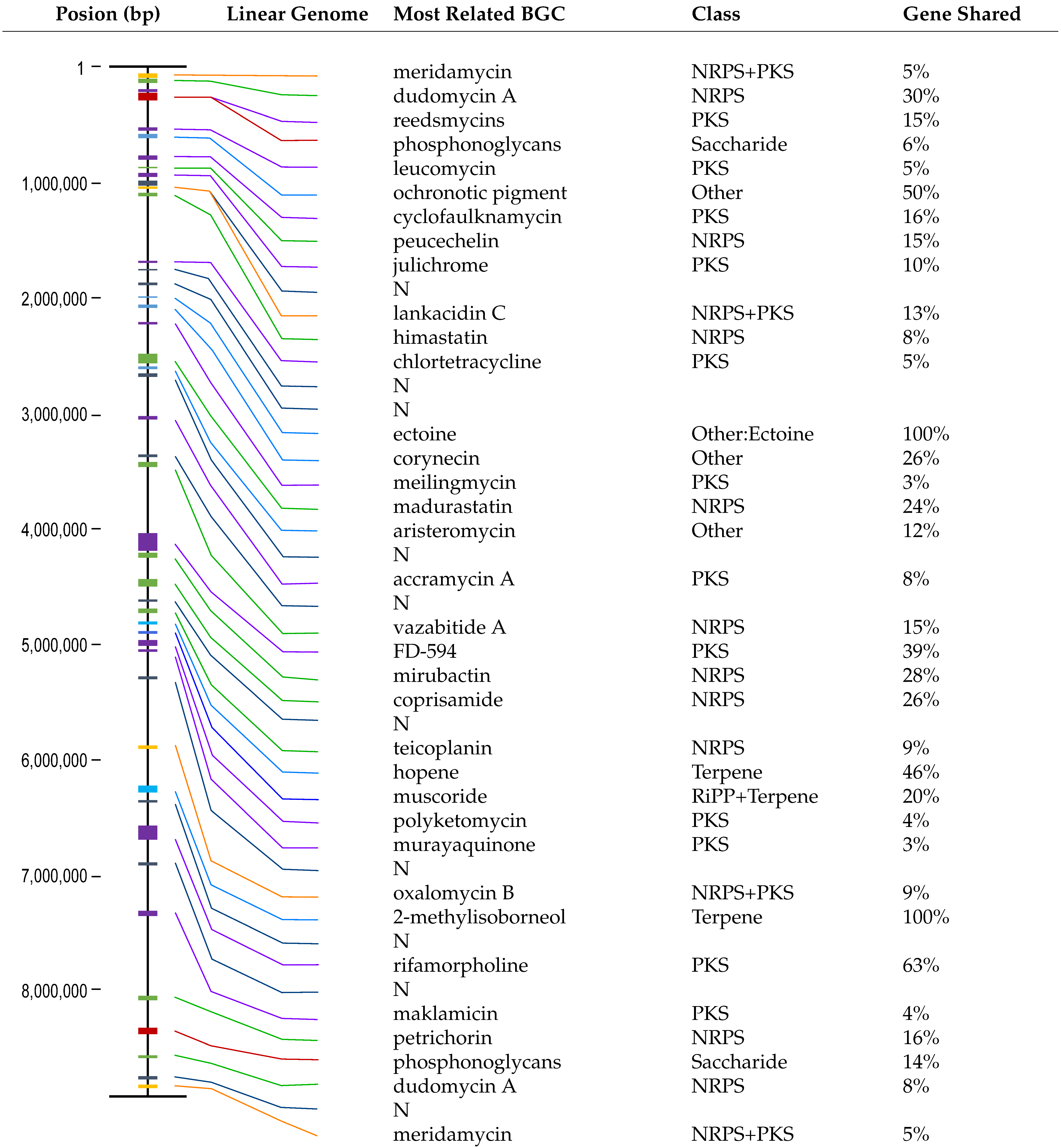
3.1. Identification and Characterization of the TRM71106 Strain
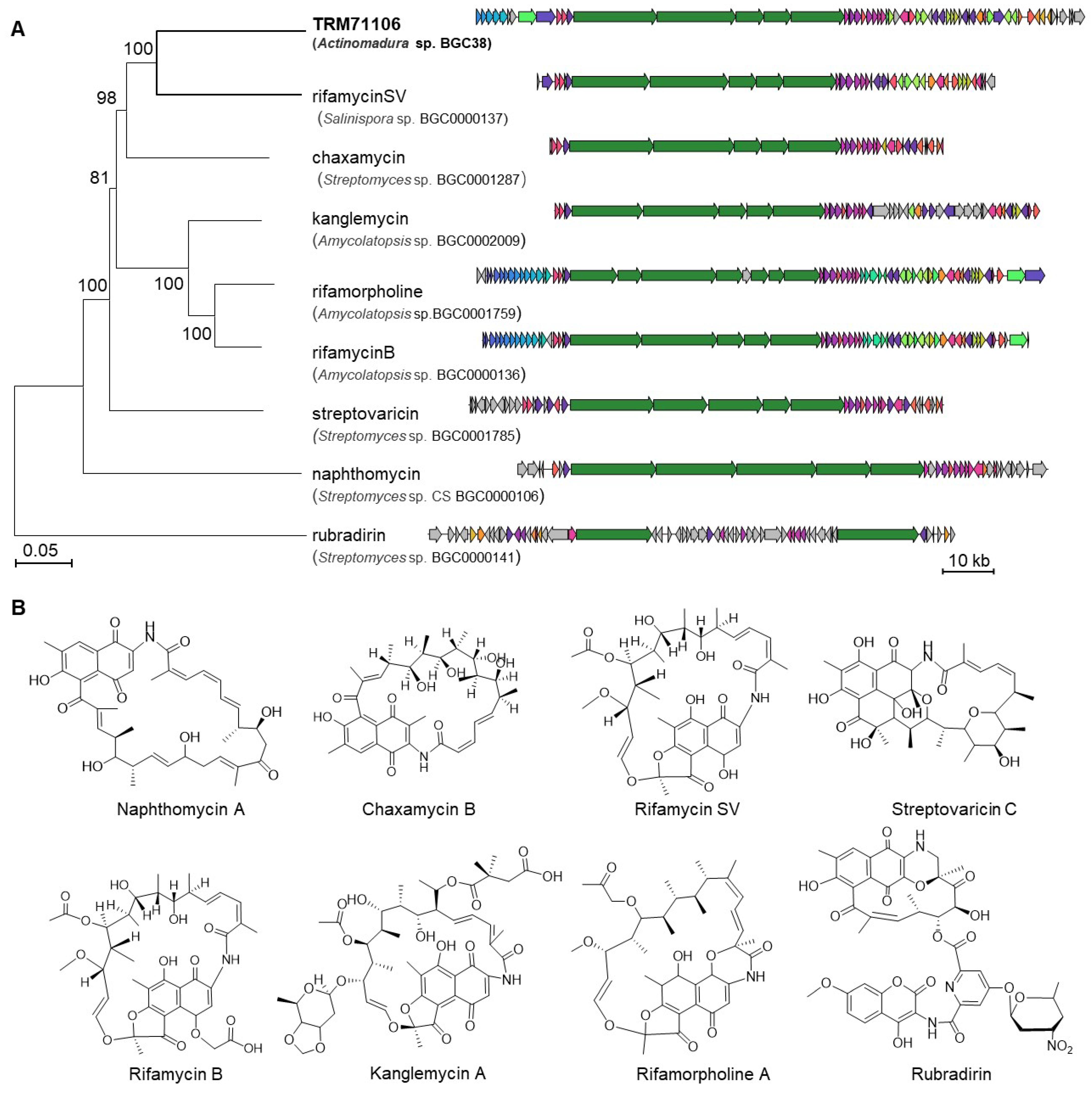
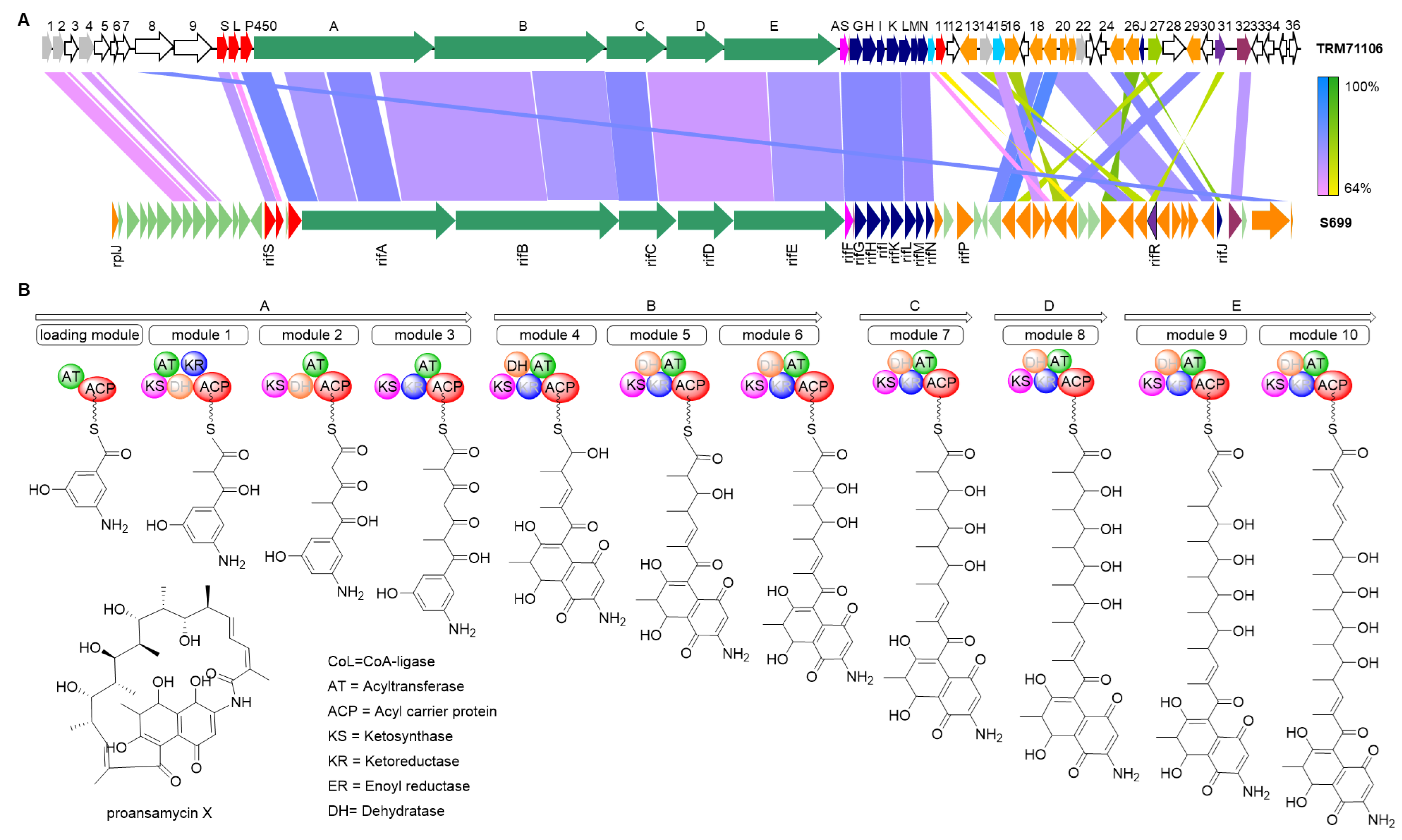
3.2. Results of Biosynthetic Gene Cluster Analysis
3.3. TRM71160-IL Srain Construction and Detection
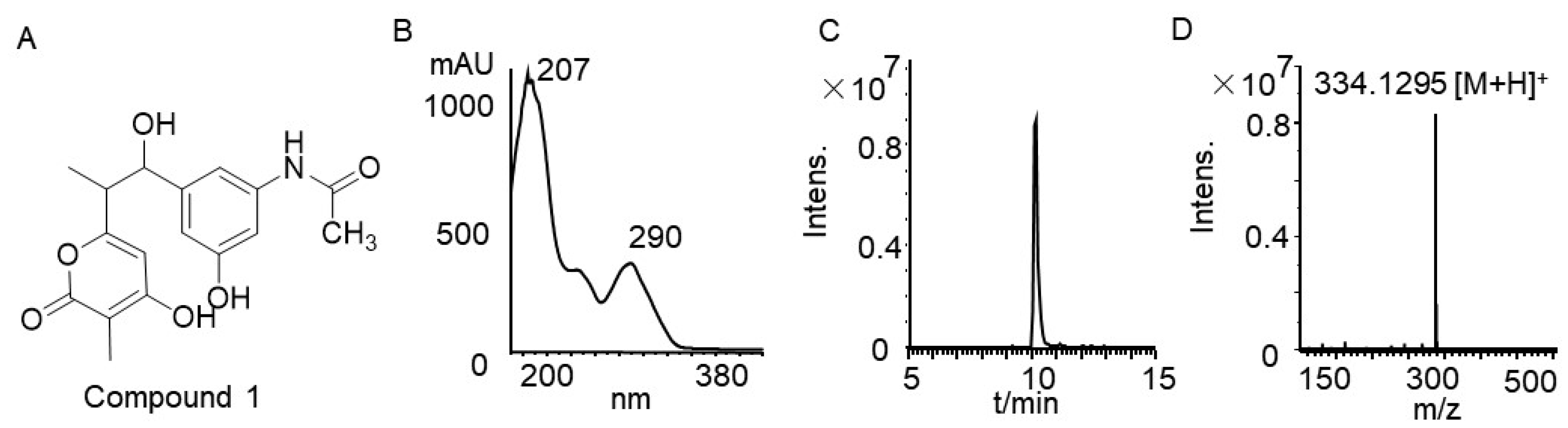
3.4. Rifamycin Produced by Strain TRM71160-IL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ortega, M.A.; van der Donk, W.A. New Insights into the Biosynthetic Logic of Ribosomally Synthesized and Post-translationally Modified Peptide Natural Products. Cell Chem. Biol. 2016, 23, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Meenakshi, S.; Hiremath, J.; Meenakshi, M.H.; Shivaveerakumar, S. Actinomycetes: Isolation, Cultivation and its Active Biomolecules. J. Pure Appl. Microbiol. 2024, 18, 118–143. [Google Scholar] [CrossRef]
- Alam, K.; Mazumder, A.; Sikdar, S.; Zhao, Y.M.; Hao, J.; Song, C.; Wang, Y.; Sarkar, R.; Islam, S.; Zhang, Y.; et al. Streptomyces: The biofactory of secondary metabolites. Front. Microbiol. 2022, 13, 968053. [Google Scholar] [CrossRef] [PubMed]
- Ezeobiora, C.E.; Igbokwe, N.H.; Amin, D.H.; Enwuru, N.V.; Okpalanwa, C.F.; Mendie, U.E. Uncovering the biodiversity and biosynthetic potentials of rare actinomycetes. Future J. Pharm. Sci. 2022, 8, 23. [Google Scholar] [CrossRef]
- Tiwari, K.; Gupta, R.K. Diversity and isolation of rare actinomycetes: An overview. Crit. Rev. Microbiol. 2013, 39, 256–294. [Google Scholar] [CrossRef]
- Bull, A.T.; Goodfellow, M. Dark, rare and inspirational microbial matter in the extremobiosphere: 16 000 m of bioprospecting campaigns. Microbiology 2019, 165, 1252–1264. [Google Scholar] [CrossRef]
- Hu, D.; Gao, C.; Sun, C.; Jin, T.; Fan, G.; Mok, K.M.; Lee, S.M.-Y. Genome-guided and mass spectrometry investigation of natural products produced by a potential new actinobacterial strain isolated from a mangrove ecosystem in Futian, Shenzhen, China. Sci. Rep. 2019, 9, 823. [Google Scholar] [CrossRef]
- Xiao, Y.S.; Zhang, B.; Zhang, M.; Guo, Z.K.; Deng, X.Z.; Shi, J.; Li, W.; Jiao, R.H.; Tan, R.X.; Ge, H.M. Rifamorpholines A-E, potential antibiotics from locust-associated actinobacteria Amycolatopsis sp. Hca4. Org. Biomol. Chem. 2017, 15, 3909–3916. [Google Scholar] [CrossRef]
- Xie, F.; Pathom-aree, W. Actinobacteria From Desert: Diversity and Biotechnological Applications. Front. Microbiol. 2021, 12, 765531. [Google Scholar] [CrossRef]
- Murtaza, M.; Abrol, V.; Nehra, E.; Choudhary, P.; Singh, S.K.; Jaglan, S. Biodiversity and Bioactive Potential of Actinomycetes from Unexplored High Altitude Regions of Kargil, India. Indian J. Microbiol. 2024, 64, 110–124. [Google Scholar] [CrossRef]
- Sivalingam, P.; Hong, K.; Pote, J.; Prabakar, K. Extreme Environment Streptomyces: Potential Sources for New Antibacterial and Anticancer Drug Leads? Int. J. Microbiol. 2019, 2019, 5283948. [Google Scholar] [CrossRef]
- Jiang, Q.D.; Yang, X.P. Sedimentological and Geochemical Composition of Aeolian Sediments in the Taklamakan Desert: Implications for Provenance and Sediment Supply Mechanisms. J. Geophys. Res.-Earth Surf. 2019, 124, 1217–1237. [Google Scholar] [CrossRef]
- Wang, T.; Li, F.; Lu, Q.; Wu, G.; Jiang, Z.; Liu, S.; Habden, X.; Razumova, E.A.; Osterman, I.A.; Sergiev, P.V.; et al. Diversity, novelty, antimicrobial activity, and new antibiotics of cultivable endophytic actinobacteria isolated from psammophytes collected from Taklamakan Desert. J. Pharm. Anal. 2021, 11, 241–250. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Couteau, C.; Luo, F.; Neveu, J.; DuBow, M.S. Bacterial diversity of surface sand samples from the Gobi and Taklamaken deserts. Microb. Ecol. 2013, 66, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, Y.; Huang, C.; Luo, Y. Recent Advances in Silent Gene Cluster Activation in Streptomyces. Front. Bioeng. Biotechnol. 2021, 9, 632230. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes—A review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Fagan, A.; Gavis, E.A.; Mousel, T.; Gallagher, M.L.; Puri, P.; Fuchs, M.; Davis, B.C.; Hylemon, P.B.; Zhou, H.; et al. The RIVET RCT: Rifamycin SV MMX improves muscle mass, physical function, and ammonia in cirrhosis and minimal encephalopathy. Hepatol. Commun. 2024, 8, e0384. [Google Scholar] [CrossRef]
- Zinyakatira, N.; Ford, N.; Cox, H. Association between HIV and acquisition of rifamycin resistance with first-line TB treatment: A systematic review and meta-analysis. BMC Infect. Dis. 2024, 24, 657. [Google Scholar] [CrossRef]
- Lindgreen, S. AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 2012, 5, 337. [Google Scholar] [CrossRef]
- Luo, C.; Tsementzi, D.; Kyrpides, N.C.; Konstantinidis, K.T. Individual genome assembly from complex community short-read metagenomic datasets. ISME J. 2012, 6, 898–901. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Son, P.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Petrov, A.I.; Kay, S.J.E.; Kalvari, I.; Howe, K.L.; Gray, K.A.; Bruford, E.A.; Kersey, P.J.; Cochrane, G.; Finn, R.D.; Bateman, A.; et al. RNAcentral: A comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 2017, 45, D128–D134. [Google Scholar] [CrossRef]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR Recognition Tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Chooi, Y.-H. clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Munoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef]
- Laureti, L.; Song, L.; Huang, S.; Corre, C.; Leblond, P.; Challis, G.L.; Aigle, B. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc. Natl. Acad. Sci. USA 2011, 108, 6258–6263. [Google Scholar] [CrossRef]
- August, P.R.; Tang, L.; Yoon, Y.J.; Ning, S.; Muller, R.; Yu, T.W.; Taylor, M.; Hoffmann, D.; Kim, C.G.; Zhang, X.H.; et al. Biosynthesis of the ansamycin antibiotic rifamycin: Deductions from the molecular analysis of the rif biosynthetic gene cluster of Amycolatopsis mediterranei S699. Chem. Biol. 1998, 5, 69–79. [Google Scholar] [CrossRef]
- Yu, T.W.; Shen, Y.; Doi-Katayama, Y.; Tang, L.; Park, C.; Moore, B.S.; Richard Hutchinson, C.; Floss, H.G. Direct Evidence That the Rifamycin Polyketide Synthase Assembles Polyketide Chains Processively. Proc. Natl. Acad. Sci. USA 1999, 96, 9051–9056. [Google Scholar] [CrossRef]
- Hong Jay Sung, J.; Choi, C.Y.; Yoon, Y.J. Premature release of polyketide intermediates by hybrid polyketide synthase in Amycolatopsis mediterranei S699. J. Microbiol. Biotechnol. 2003, 13, 613–619. [Google Scholar]
- Huang, H.; Wu, X.; Yi, S.; Zhou, Z.; Zhu, J.; Fang, Z.; Yue, J.; Bao, S.J.A.V.L. Rifamycin S and its geometric isomer produced by a newly found actinomycete, Micromonospora rifamycinica. Antonie Van. Leeuwenhoek 2009, 95, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, N.; Iwasaki, S.; Yazawa, K.; Mikami, Y.; Maeda, A. Inactivated products of rifampicin by pathogenic Nocardia spp.: Structures of glycosylated and phosphorylated metabolites of rifampicin and 3-formylrifamycin SV. J. Antibiot. 1993, 46, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Xu, Z.; Guo, Z.; Hindra; Ma, M.; Yang, D.; Zhou, H.; Gansemans, Y.; Zhu, X.; Huang, Y.; et al. Discovery of the leinamycin family of natural products by mining actinobacterial genomes. Proc. Natl. Acad. Sci. USA 2017, 114, E11131–E11140. [Google Scholar] [CrossRef]
- Sensi, P.; Margalith, P.; Timbal, M.T. Rifomycin, a new antibiotic; preliminary report. Il Farm. Ed. Sci. 1959, 14, 146–147. [Google Scholar]
- Lucchesi, M.; Bancale, A.; Pallotta, G.; Termine, A. 1st clinical observations in the tuberculosis field with rifamycin SV administered by venous perfusion. Ann. Dell’istituto Carlo Forlanini 1962, 22, 441–469. [Google Scholar]
- Maggi, N.; Pasqualucci, C.R.; Ballotta, R.; Sensi, P. Rifampicin: A New Orally Active Rifamycin. Chemotherapy 1966, 11, 285–292. [Google Scholar] [CrossRef]
- Wei, F.; Wang, Z.; Lu, C.; Li, Y.; Zhu, J.; Wang, H.; Shen, Y. Targeted Discovery of Pentaketide Ansamycin Aminoansamycins A-G. Org. Lett. 2019, 21, 7818–7822. [Google Scholar] [CrossRef]
- Skrzypczak, N.; Przybylski, P. Structural diversity and biological relevance of benzenoid and atypical ansamycins and their congeners. Nat. Prod. Rep. 2022, 39, 1678–1704. [Google Scholar] [CrossRef]
- Phelan, V.V. Feature-Based Molecular Networking for Metabolite Annotation. In Computational Methods and Data Analysis for Metabolomics; Li, S., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2020; Volume 2104, pp. 227–243. [Google Scholar]
- Tong, Y.; Whitford, C.M.; Blin, K.; Jorgensen, T.S.; Weber, T.; Lee, S.Y. CRISPR-Cas9, CRISPRi and CRISPR-BEST-mediated genetic manipulation in streptomycetes. Nat. Protoc. 2020, 15, 2470–2502. [Google Scholar] [CrossRef]
- Kieser, T.; Bibb, M.J.; Buttner, M.J.; Chater, K.F.; Hopwood, D.A. Practical Streptomyces Genetics; John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
- Bierman, M.; Logan, R.; O’Brien, K.; Seno, E.T.; Nagaraja Rao, R.; Schoner, B.E. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 1992, 116, 43–49. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, X.; Liu, Z.; Luo, X.; Xia, Z.; Wan, C.; Wang, H.; Zhang, L. Genome Mining Reveals Rifamycin Biosynthesis in a Taklamakan Desert Actinomycete. Microorganisms 2025, 13, 1068. https://doi.org/10.3390/microorganisms13051068
Luo X, Liu Z, Luo X, Xia Z, Wan C, Wang H, Zhang L. Genome Mining Reveals Rifamycin Biosynthesis in a Taklamakan Desert Actinomycete. Microorganisms. 2025; 13(5):1068. https://doi.org/10.3390/microorganisms13051068
Chicago/Turabian StyleLuo, Xinrong, Zhanwen Liu, Xiaoxia Luo, Zhanfeng Xia, Chuanxing Wan, Haoxin Wang, and Lili Zhang. 2025. "Genome Mining Reveals Rifamycin Biosynthesis in a Taklamakan Desert Actinomycete" Microorganisms 13, no. 5: 1068. https://doi.org/10.3390/microorganisms13051068
APA StyleLuo, X., Liu, Z., Luo, X., Xia, Z., Wan, C., Wang, H., & Zhang, L. (2025). Genome Mining Reveals Rifamycin Biosynthesis in a Taklamakan Desert Actinomycete. Microorganisms, 13(5), 1068. https://doi.org/10.3390/microorganisms13051068